Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Spinal muscular atrophy
Last reviewed: 29.06.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Ang spinal muscular atrophy ay hindi isang solong nosologic unit, ngunit isang buong grupo ng mga clinically at genetically heterogenous hereditary pathologies na pinukaw ng pagtaas ng mga proseso ng pagkabulok ng mga motoneuron ng anterior spinal horns. Ang termino ay sumasaklaw sa iba't ibang variant ng genetically determined peripheral paresis at muscular atrophy na nagreresulta mula sa pagkabulok ng mga spinal motor neuron at/o brainstem. Ang pinakakaraniwang sanhi ng problema ay isang autosomal recessive mutation sa mahabang q-balikat ng ikalimang chromosome. Ang paggamot ay hindi tiyak, na naglalayong mapabuti ang trophicity ng nervous tissue at magbigay ng palliative na suporta upang mapabuti ang kalidad ng buhay. [ 1 ]
Epidemiology
Ang spinal muscular atrophy ay nangyayari sa isang kaso bawat 6,000 hanggang 10,000 bagong panganak (ayon sa American Journal of Medical Genetics 2002).
Ang prevalence ng SMN gene exon 7 deletion carriers ay 1:50 tao.
Ang Bulbo-spinal muscular atrophy (Kennedy syndrome) ay nangyayari sa isang bata sa 50,000 at ito ang pinakakaraniwang pang-adultong uri ng spinal amyotrophy.
Ito ay nabanggit na kalahati ng mga bata na may sakit na ito ay hindi nagtagumpay sa dalawang taon na panahon ng kaligtasan.
Ang patolohiya ay minana ayon sa prinsipyo ng autosomal recessive. Kadalasan, ang bawat magulang ng isang may sakit na bata ay isang carrier ng isang kopya ng mutated gene. Dahil ang mutation ay binabayaran ng pagkakaroon ng pangalawang "normal" na kopya ng gene, ang mga magulang ay walang mga pagpapakita ng spinal muscular atrophy. Ang type 2 na patolohiya ay karaniwang hindi nagmamana ng karagdagang kopya mula sa magulang. Ang problema ay nangyayari dahil sa isang aksidenteng pagkabigo sa panahon ng pagbuo ng mga selula ng mikrobyo, o direkta sa oras ng pagpapabunga. Sa spinal muscular atrophy ng unang uri, ang kusang pag-unlad ng sakit ay nangyayari sa 2% lamang ng mga kaso (sa sitwasyong ito, ang carrier ay isa lamang sa mga magulang). [ 2 ]
Mga sanhi ng spinal muscular atrophy
Ang pangunahing sanhi ng pagkasayang ng kalamnan ng gulugod ay isang mutation ng gene na responsable para sa paggawa ng protina ng SMN na naisalokal sa chromosome 5q. Ang karamdamang ito ay higit na nagdudulot ng unti-unting pagkamatay ng mga selula ng motor nerve sa mga anterior horn ng spinal cord at brain stem. Bilang resulta ng mga prosesong ito, bumababa ang tono ng musculature, pagkasayang ng respiratory, pharyngeal, facial at skeletal na kalamnan. Ang nangingibabaw na uri ng pamana ng mga pediatric form ng spinal muscular atrophy ay autosomal recessive, na nagpapahiwatig ng sabay-sabay na pagdadala ng mga may sira na gene ng parehong mga magulang. Tulad ng para sa type IV na patolohiya (pang-adultong anyo), mayroong isang link sa X chromosome, kaya ang mga lalaki lamang ang apektado.
Ang pag-unlad ng pagkasayang ng kalamnan ng gulugod ay batay sa pagtaas ng mga proseso ng pagkabulok at pagkamatay ng mga neuron ng motor ng mga sungay ng spinal anterior, pinsala sa nuclei ng stem ng utak. Ang mga pagbabago sa patolohiya ay pinakamatindi sa mga zone ng cervical at lumbar thickening. Ang cellular number ay nabawasan sa isang minimum, ang kapalit ng connective tissue ay nangyayari, na dahil sa pagkabigo ng cell death program - ang tinatawag na apoptosis. Ang pagbabago ay nakakaapekto sa mga istruktura ng motor nuclei ng cranial nerves, anterior roots, motor nerves. Mayroong isang klinika ng neurogenic fascicular atrophy. Sa isang matagal na kurso ng sakit sa isang huling yugto ng nag-uugnay na tissue overgrowth nangyayari.
Ang hitsura ng kaukulang klinikal na larawan ay nauugnay sa isang kakulangan ng protina ng SMN, na nakakaimpluwensya sa matagumpay na pag-andar ng mga cell nerve ng motor sa mga anterior spinal horn. Ang kakulangan sa protina bilang isa sa mga link sa pagbuo ng spinal muscular atrophy ay natuklasan sa pagtatapos ng XX century. Laban sa background ng pinsala sa motoneuron, ang innervation ng mga kalamnan ng kalansay (pangunahin ang mga proximal na seksyon) ay may kapansanan. [ 3 ]
Mga kadahilanan ng peligro
Ang pagkakaiba-iba ng mga klinikal na anyo ng spinal muscular atrophy 5q ay ipinaliwanag sa pamamagitan ng pagkakaroon ng ilang partikular na salik sa pagbabago na maaaring nahahati sa dalawang kategorya: ang mga nakakaapekto at ang mga hindi nakakaapekto sa marka ng protina ng SMN.
- Sa kasalukuyan, ang SMN2 gene ay itinuturing na pangunahing kadahilanan sa pagbuo ng spinal muscular atrophy: mas maraming kopya ng SMN2 gene, mas mababa ang intensity ng mga sintomas ng sakit. Ang pangalawang kadahilanan, na direktang nauugnay sa centromeric na kopya ng SMN gene, ay isang 1-nucleotide substitution c.859G>C sa exon 7 ng SMN2 gene, na humahantong sa pagbuo ng isang bagong enhancer-binding splice site: ang resulta ay ang pagsasama ng exon 7 sa transcript mula sa SMN2 gene. Ang pagkakaiba-iba na ito ay nauugnay sa isang pagtaas sa antas ng dugo ng full-length na protina ng SMN sa mga pasyente na may spinal amyotrophy ng pangalawa o pangatlong uri.
Iba pang mga salik na nakakaapekto sa bilang ng mga SMN:
- Splicing-regulatory factors (Tra2β - induces exon skipping of exon 7, SF2/ASF - increases exon 7 inclusion, hnRNPA1 - suppresses exon 7 inclusion of the SMN2 gene).
- Mga salik sa regulasyon ng transkripsyon (CREB1 - pinapataas ang transkripsyon ng SMN, STAT3 - pinapaboran ang paglaki ng axon, IRF1 - pinapataas ang numero ng SMN, PRL - pinapataas ang habang-buhay sa mga malubhang yugto).
- MRNA stabilizing factor (U1A-binabawasan ang SMN, HuR/p38).
- Mga salik na nakakaapekto sa post-translational modification (RCA - pinipigilan ang pagkasira ng SMN, GSK3 - pinapataas ang kaligtasan).
- Exogenous na mga kadahilanan (gutom, hypoxia, oxidative stress).
Ang mga epekto ng mga salik sa itaas ay natutukoy nang nakararami sa vitro.
- Mga salik na hindi nauugnay sa SMN gene - sa partikular, mga protina na nag-o-optimize ng endocytosis sa mga synapses (laminin 3, coronin, neurocalcin delta, calcium-neurin-like protein).
Ang karagdagang pansin ay binabayaran sa DNA methylation, ang pinaka-matatag na pagbabago na nakakaapekto sa likas na katangian ng pagpapahayag ng gene. Ang methylation ng isang pangkat ng mga gene na posibleng kasangkot sa mga pathogenetic na proseso ay natagpuang nauugnay sa kalubhaan ng spinal muscular atrophy. [ 4 ]
Pathogenesis
Ang spinal muscular atrophy ay isang genetic pathology kung saan ang alinman sa mga uri ng inheritance - parehong autosomal dominant at autosomal recessive o X-linked - ay likas. Kadalasan ay pinag-uusapan natin ang tungkol sa maagang pagkabata ng autosomal recessive na patolohiya. Ang responsibilidad para sa pagbuo ng naturang spinal amyotrophy ay ang SMN gene, na naisalokal sa locus 5q13. Ang pagtanggal ng exon 7 sa SMN gene ay nagreresulta sa patolohiya na may posibleng paglahok ng mga kalapit na gen p44 at NAIP.
Ang SNM genome ay nag-encode ng isang protina na kinabibilangan ng 294 amino acid at may MM na ~38 kDa. Ang protina ay may mga sumusunod na pag-andar:
- Ay bahagi ng RNA-protein complex;
- Nakikilahok sa pagbuo ng spliceosome site na nag-catalyze ng pre-RNA splicing;
- Kasangkot sa mga prosesong kumokontrol sa produksyon ng protina at mga isoform ng protina;
- Nagbibigay ng axonal transport ng mRNA;
- Pinapaboran ang paglaki ng nerve cell at nagbibigay ng neuromuscular communication.
Ang ilang uri ng mga gene ng SMN ay kilala:
- Telomeric SMnt (SMN1);
- Centromeric SMnc (SMN2).
Ang karamihan sa mga kaso ng spinal muscular atrophy ay dahil sa mga pagbabago sa SMN1 gene.
Ang Kennedy spinal muscular atrophy ay may linkage sa Xq12 locus na naglalaman ng NR3C3 gene, na nag-encode ng androgen receptor protein. Mayroon itong X-linked inheritance variant. Kapag ang bilang ng CAG ay umuulit sa isang gene exon ay tumaas, ang patolohiya ay bubuo.
Ang pagsugpo sa produksyon ng protina ng SNM ay sinamahan ng mga sumusunod na pagbabago:
- Dahil sa kapansanan sa koordinasyon ng axon, ang labis na pagsanga ng mga axon ay nangyayari;
- Ang paglaki ng mga axon ay bumabagal at ang kanilang laki ay bumababa;
- Mayroong hindi tamang pag-cluster ng mga channel ng calcium sa growth cone;
- Ang hindi regular na presympathetic na mga terminal ng motor nerve cell axons ay nabuo.
Ang spinal cord ay nagsisimulang aktibong mawalan ng mga neuron ng motor sa mga anterior na sungay, na nagiging sanhi ng pag-unlad ng pagkasayang ng mga proximal na kalamnan ng paa. [ 5 ]
Mga sintomas ng spinal muscular atrophy
Symptomatology ng spinal muscular atrophy na si Werdnig-Hoffman ay madalas na nag-debut sa panahon ng bagong panganak at hanggang anim na buwan, na ipinakita ng sindrom ng isang "tamad" na sanggol. Ang dibdib na hugis kampana, matinding hypotonia, kakulangan ng reflexes, pagkibot ng kalamnan ng dila at paghinga sa paghinga ay napansin. Ang mga may sakit na sanggol ay mas madalas na namamatay bago umabot sa edad na dalawang taon: ang nakamamatay na kinalabasan ay dahil sa pagtaas ng respiratory failure laban sa background ng pagsunod sa mga nakakahawang proseso.
Ang intermediate form ng spinal muscular atrophy ng pangalawang uri ay napansin mula sa edad na anim na buwan. Bilang karagdagan sa sindrom ng isang "tamad" na bata, mayroong mababang presyon ng dugo, kakulangan ng mga reflexes, mga sakit sa paghinga at pagkibot ng dila. Kahit na ang mga bata ay nakakaupo, maraming contracture ng malalaking joints ang bubuo.
Ang Kugelberg-Wielander spinal muscular atrophy ay nagsisimula din sa maagang pagkabata, kung saan ang mga bata ay nakakagalaw nang nakapag-iisa. Mayroong panghihina ng iliac, quadriceps at adductor muscles, mababang presyon ng dugo, nabawasan ang reflexes at pagkibot ng dila. Maraming mga pasyente ang nawalan ng kakayahang gumalaw (maglakad) nang nakapag-iisa sa paglipas ng mga taon.
Ang spinal muscular atrophy type 4 ay nagsisimula sa mas matandang edad. Ito ay nailalarawan sa pamamagitan ng mabagal na pag-unlad at medyo benign prognosis. [ 6 ]
Ang pagkasayang ni Kennedy ay madalas na nagpapakita ng sarili sa gitna ng edad (karaniwang maaaring mag-debut sa mga pasyente na 15-60 taong gulang). Kasama sa symptomatology ang pananakit at panghihina ng kalamnan, gynecomastia, distal na kahinaan, pagkahilo, pagkibot ng dila at pagkasayang. Ang mga palatandaan ng bulbar dysfunction ay naroroon:
- Kahirapan sa paglunok;
- Aspirasyon;
- Paghina ng mga kalamnan ng masticatory;
- Dysarthria;
- Panginginig ng postural at motor sa mga kamay.
Ang mga unang palatandaan ng kakulangan sa androgen:
- Gynecomastia (sa halos 60% ng mga pasyente), kadalasang walang simetrya;
- Pagkasira ng sexual function (oligospermia, testicular atrophy, erectile dysfunction).
Mga unang palatandaan
Ang spinal amyotrophy ay ipinakita sa pamamagitan ng kahinaan ng mga kalamnan at pangkalahatang kawalan ng lakas. Ang lahat ng pandama at intelektwal na kakayahan ay hindi apektado.
Mga pangunahing indeks ng neuromuscular pathology:
- Musculature "tamad", weakened, laxity at laxity ng mga kalamnan ay nabanggit;
- Ang tono ng kalamnan ay mababa, ang mga tendon reflexes ay pinaliit o wala;
- Normal o wala ang mga plantar reflexes;
- Ang mga maikling twitch ng mga indibidwal na grupo ng kalamnan ay nabanggit (maaaring makita sa ilalim ng balat, sa dila);
- May mga palatandaan ng pagkasayang ng kalamnan.
Ang Werdnig-Hoffman syndrome ay ipinahayag sa pamamagitan ng binibigkas na hypotonia ng mga kalamnan, pangkalahatang pagkahilo, kawalan ng kakayahan ng bata na hawakan ang ulo, tumalikod at umupo. Kapag sinusubukang suportahan ang sanggol sa lugar ng tiyan sa isang suspendido na estado, ang katawan ay tila "lumubog". Ang pag-ubo, paglunok at pagsuso ng reflex ay hindi kasiya-siya, ang pagkain ay madalas na pumapasok sa respiratory tract, ang paghinga ay may problema. Maaaring may joint distortion na nauugnay sa intrauterine hypotonia. Ang anamnestic na impormasyon na nakolekta sa panahon ng pagbubuntis ay madalas na nagpapahiwatig ng mababang aktibidad ng pangsanggol.
Mga pangunahing palatandaan ng spinal muscular atrophy type I:
- Malubhang pagkaantala sa pag-unlad ng motor;
- Mabilis na pagsisimula ng joint contractures at thoracic curvature;
- Ang pagtaas ng mga sakit sa paghinga at bulbar, mga problema sa paglunok (parehong pagkain at laway) at paglabas ng plema;
- Nadagdagang panganib ng pamamaga ng aspirasyon;
- Impeksyon, progresibong pagkabigo sa paghinga.
Ang spinal muscular atrophy type II ay ipinakita sa pamamagitan ng isang malinaw na pagsugpo sa pag-unlad ng motor. Bagaman maraming mga pasyente ang maaaring umupo nang walang tulong, at kung minsan ay gumagapang at tumayo, ang mga kakayahan na ito ay kadalasang nawawala sa paglipas ng panahon. Ang mga panginginig ng daliri, mga distortion ng kalamnan at kasukasuan (buto), at mga problema sa paghinga ay nabanggit. Posibleng calf pseudohypertrophy.
Ang mga pangunahing tampok ng type II patolohiya:
- Mga pagkaantala sa pag-unlad, kabilang ang paghinto at pagbaligtad sa pagbuo ng mga nakuha nang mga kasanayan at kakayahan;
- Ang pagtaas ng kahinaan ng mga intercostal na kalamnan;
- Superficiality ng diaphragmatic breathing, weakened cough reflex, unti-unting paglala ng respiratory failure;
- Curvature ng thorax at spinal column, contractures.
Sa Kugelberg-Wielander syndrome, ang mga pagpapakita ay mas banayad, dahan-dahang umuunlad. Ang pasyente ay nakakagalaw, ngunit may mga problema sa jogging o pag-akyat ng hagdan. Ang mga naantalang sintomas ay kadalasang kinabibilangan ng kahirapan sa paglunok at pagnguya.
Ang spinal muscular atrophy type IV ay nagpapakita ng sarili sa mas matanda (adult) na edad at nailalarawan sa pamamagitan ng pinaka "banayad" at kanais-nais na kurso. Ang mga pangunahing palatandaan: unti-unting pagkawala ng kakayahang lumipat. [ 7 ]
Mga Form
Ang spinal muscular atrophy ay bahagi ng isang pangkat ng mga namamana na pathologies na nailalarawan sa pamamagitan ng mga degenerative na pagbabago, pagkamatay ng mga selula ng motor nerve ng anterior spinal horns at, madalas, ang motor nuclei ng brainstem. Ang proseso ay maaaring ipakilala ang sarili sa iba't ibang panahon ng buhay, ang klinikal na larawan ay hindi palaging pareho. Ang mga uri ng mana at kurso ay maaari ding magkaiba.
Ang pediatric spinal muscular atrophy ay unang inilarawan noong huling bahagi ng ika-19 na siglo. Sa kalagitnaan ng ika-20 siglo, ang mga pangunahing anyo ng sakit ay nakilala:
- Congenital (nagpapakita mismo halos kaagad pagkatapos ng kapanganakan ng sanggol);
- Maagang infantile form (nagaganap laban sa background ng nakaraang normal na pag-unlad ng sanggol);
- Late infantile form (nagpapakita ng sarili simula sa edad na 2 at mas matanda).
Pinagsasama ng ilang mga espesyalista ang pangalawa at pangatlong anyo sa isang pediatric na uri ng spinal amyotrophy.
Karaniwang tinatanggap na hatiin ang patolohiya sa pediatric at adult. Ang spinal muscular atrophy sa mga bata ay inuri sa maaga (na may pasinaya sa unang ilang buwan pagkatapos ng kapanganakan ng bata), huli at nagdadalaga (nagbibinata, o juvenile). Ang mga sindrom na kadalasang nasasangkot ay:
- Pagkasayang ng Werdnig-Hoffman;
- Ang form na Kugelberg-Wielander;
- Talamak na infantile spinal muscular atrophy;
- Vialetto-van Lare syndrome (uri ng bulbospinal na walang pandinig);
- Fazio-Londe syndrome.
Ang pang-adultong spinal muscular atrophy ay nagsisimula sa edad na 16 na taon at hanggang humigit-kumulang 60 taong gulang, na nakikilala sa pamamagitan ng medyo benign na klinika at pagbabala. Kasama sa mga pathology ng may sapat na gulang ang:
- Ang bulbospinal atrophy ni Kennedy;
- Scapuloperoneal atrophy;
- Mga anyo ng facial-lap-shoulder at oculo-pharyngeal;
- Distal spinal atrophy;
- Monomelic spinal atrophy.
Hiwalay na hiwalay ang hiwalay at pinagsamang pagkasayang ng gulugod. Ang nakahiwalay na patolohiya ay nailalarawan sa pamamagitan ng pamamayani ng pinsala sa mga spinal motor neuron (na kadalasang ang tanging tanda ng problema). Ang pinagsamang patolohiya ay bihira at kumakatawan sa isang kumplikado ng mga neurological at somatic disorder. May mga paglalarawan ng mga kaso ng pinagsamang sindrom na may congenital coronary malformations, kakulangan ng auditory function, oligophrenia, cerebellar hypoplasia.
Ang spinal muscular atrophy sa mga matatanda ay pinaka-karaniwang kinakatawan ng Kennedy bulbospinal amyotrophy. Ang patolohiya na ito ay minana ng recessively X-linked. Ang kurso ng sakit ay mabagal, medyo benign. Nagsisimula ito sa pagkasayang ng proximal musculature ng lower extremities. Posibleng panginginig ng mga kamay, ulo. Kasabay nito, ang mga problema sa endocrine ay napansin din: testicular atrophy, gynecomastia, diabetes mellitus. Sa kabila nito, sa mga matatanda, ang patolohiya ay nagpapatuloy sa mas banayad na anyo kaysa sa mga bata.
Isang variant ng spinal muscular atrophy. |
Ang pasinaya ng patolohiya |
Nakikitang problema |
Edad ng kamatayan |
Mga katangian ng symptomatology |
Spinal muscular atrophy type 1 (iba pang pangalan Verding-Hoffman spinal muscular atrophy) |
Mula sa kapanganakan hanggang anim na buwan |
Hindi makaupo ang sanggol |
Hanggang dalawang taon |
Matinding panghihina ng kalamnan, hypotonia, problema sa paghawak sa ulo, kapansanan sa pag-iyak at pag-ubo, mga problema sa paglunok at paglalaway, pag-unlad ng respiratory failure at aspiration pneumonia |
Uri ng spinal muscular atrophy 2 |
Anim na buwan hanggang isa at kalahating taon |
Hindi makatayo ang sanggol |
Mahigit dalawang taon |
Paghina ng motor, kakulangan sa timbang, panghihina ng ubo, panginginig ng kamay, kurbada ng gulugod, mga contracture |
Spinal muscular atrophy type 3 (iba pang pangalan Kugelberg-Welander spinal muscular atrophy) |
Pagkatapos ng isang taon at kalahati. |
Sa simula ay maaaring tumayo at maglakad, ngunit sa isang tiyak na edad ang kakayahang ito ay maaaring mawala |
Sa pagtanda. |
Nanghina ang mga kalamnan, contracture, joint hypermobility |
Uri ng spinal muscular atrophy 4. |
Pagbibinata o pagtanda |
Sa simula ay maaaring tumayo at maglakad, ngunit sa isang tiyak na edad ang kakayahang ito ay maaaring mawala |
Sa pagtanda. |
Ang pagtaas ng proximal na panghihina ng kalamnan, pagbaba ng tendon reflexes, pagkibot ng kalamnan (fasciculations) |
Tungkol sa distal spinal atrophy ay sinabi sa kaso ng mga sugat ng motor nerve cells ng spinal cord, na nagpapaloob sa ibabang bahagi ng katawan. Ang mga katangian ng palatandaan ng naturang patolohiya ay:
- Pagkasayang ng mga kalamnan ng hita;
- Panghihina sa mga tuhod, mga extensor ng paa, at mga kalamnan ng hip adductor.
Walang pagbabago sa tendon reflexes.
Ang distal na spinal muscular atrophy ay kinakatawan ng dalawang allelic variation na may magkakapatong na phenotype:
- Scapulo-perineal spinal muscular atrophy;
- Hereditary motor-sensory neuropathy ng Charcot-Marie-Tooth type 2C.
Ang proximal spinal muscular atrophy 5q ay nailalarawan sa pamamagitan ng pagtaas ng symptomatology ng flaccid paralysis at muscular atrophy, na dahil sa mga degenerative na pagbabago sa mga alpha motor neuron ng anterior spinal horns. Ang congenital disease na may postpartum asphyxia ay ang pinaka-malubhang anyo: mula sa sandaling ipinanganak ang sanggol, halos wala na ang aktibidad ng motor, may mga contracture, paglunok at mga problema sa paghinga. Sa karamihan ng mga kaso, ang gayong bata ay namamatay.
Mga komplikasyon at mga kahihinatnan
Ang karagdagang pag-unlad ng spinal amyotrophy ay humahantong sa kahinaan at pagbawas ng mass ng kalamnan ng mga limbs (lalo na sa mga binti). Ang sanggol sa una ay wala o unti-unting nawalan ng nakuha na mga kasanayan - iyon ay, nawalan ng kakayahang maglakad, umupo nang walang suporta. Ang aktibidad ng motor ng mga upper limbs ay bumababa, ang mga joints ay nagiging matigas, sa paglipas ng panahon ang contractures ay nakakabit, at ang spinal column ay nagiging curved.
Upang mapanatili ang mga kakayahan ng motor hangga't maaari at maiwasan ang pag-unlad ng mga komplikasyon, inirerekumenda:
- Magsanay ng tamang postura ng katawan (anti-gravity position), sa kama at kapag nakaupo, naglalakad, atbp..;
- Regular na physical therapy, stretching exercises, masahe, physiotherapy, anuman ang uri ng spinal muscular atrophy;
- Gumamit ng mga espesyal na kama, upuan (wheelchairs), kutson at unan;
- Pumili at gumamit ng supportive orthotics, corsets;
- Magsanay ng hydrotherapy at kinesiotherapy, na may kanais-nais na epekto sa respiratory, musculoskeletal at digestive apparatus, nervous at cardiovascular system;
- Magsagawa ng mga regular na diagnostic check-up, kabilang ang mga klinikal na pagsusuri, spinal at pelvic radiographs;
- Sistematikong kumunsulta sa isang physiotherapist at orthopedist na may karanasan sa pagtatrabaho sa mga katulad na pasyente;
- Ayusin ang mga corset, orthoses, orthopedic device, wheelchair, atbp. Depende sa dynamics.
Ang mga tagapag-alaga ng isang pasyente na may spinal muscular atrophy ay dapat na pamilyar:
- Gamit ang mga pangunahing kaalaman sa ligtas na pag-uugali, physiotherapy, masahe, physical therapy;
- Gamit ang mga patakaran ng pagpapanatili ng independiyenteng aktibidad ng pasyente, paggamit ng mga orthopedic device;
- Sa mga tuntunin ng pangangalaga, kalinisan.
Ang spinal amyotrophy ay madalas na kumplikado sa pamamagitan ng kapansanan sa pagnguya, paglunok at pagpapadaloy ng pagkain, na nagbabanta sa aspirasyon at pag-unlad ng aspiration pamamaga ng mga baga o sagabal ng respiratory tract, na pinaka-katangian ng patolohiya ng unang uri. Ang mga problema sa paglunok ay pinatutunayan ng mga sintomas tulad ng makabuluhan at patuloy na pagpapahaba ng panahon ng pagkain, pag-aatubili na kumain, pagkaing nahuhulog sa bibig, regular na pagbuga, at lumalalang pagbaba ng timbang.
Ang mga karamdaman ng digestive motility ay nagpapakita ng kanilang sarili ng paninigas ng dumi, mahinang peristalsis, matagal na pananatili ng pagkain sa tiyan (gastric stasis), ang pagbuo ng gastroesophageal reflux. Upang maiwasan ang mga naturang komplikasyon, kinakailangan:
- Subaybayan ang tamang posisyon ng pasyente habang kumakain;
- Kung kinakailangan, gumamit ng gastric tube o gastrostomy upang matiyak ang sapat na likido at nutrient na paggamit at mabawasan ang panganib ng aspirasyon;
- Sumunod sa mga alituntunin ng paghahanda ng pagkain at inumin, panoorin ang kanilang pagkakapare-pareho, at ang dalas ng mga pagkain;
- Depende sa reseta ng doktor, gumamit ng gamot, masahe, physiotherapy, atbp.
Ang isa sa mga pinaka-seryosong komplikasyon ng spinal amyotrophy ay ang dysfunction ng respiratory system na nauugnay sa kahinaan ng mga kalamnan sa paghinga. Ang mga karamdaman sa paghinga ay maaaring nakamamatay, kapwa sa mga sanggol na may type 1 na patolohiya at sa mga pasyenteng nagdadalaga at may sapat na gulang na may type 2 o 3 na sakit. Ang mga pangunahing problema ay ang mga sumusunod:
- Ang ubo reflex ay nabalisa, may mga problema sa expectoration ng plema mula sa respiratory tract;
- Ang pagtaas ng kakulangan sa dami ng hangin na pumapasok sa mga baga, may kapansanan sa paglabas ng carbon dioxide mula sa mga baga;
- Pinapangit ang dibdib, pinipiga at pinapa-deform ang mga baga;
- Mga nakakahawang proseso sa anyo ng bronchopneumonia.
Upang maiwasan ang ganitong mga komplikasyon, ang mga pasyente ay madalas na inirerekomenda na magsagawa ng mga ehersisyo sa paghinga gamit ang isang Ambu bag. [ 9 ]
Diagnostics ng spinal muscular atrophy
Sa mga pasyente na may pinaghihinalaang spinal amyotrophy, ang mga pagsisiyasat tulad ng mga ito ay may diagnostic value:
- kimika ng dugo;
- Pagsusuri ng genetic na DNA;
- Electroneuromyography.
Kabilang sa mga karagdagang pamamaraan, posibleng magtalaga ng biopsy ng mga fibers ng kalamnan, ultrasound at resonance imaging ng musculature at utak.
Ang mga pagsusuri sa dugo ay maaaring magpahiwatig na ang creatine phosphokinase ay physiologically normal, ngunit sa ilang mga kaso maaari itong tumaas sa humigit-kumulang 2.5 beses.
Ang electroneuromyogram ay nagpapakita ng mga pagbabago dahil sa pagkawala ng motor spinal neurons. Natutukoy ito sa pamamagitan ng pagbaba ng amplitude ng interference curve, ang paglitaw ng mga spontaneous active potentials, na mga fibrillation at fascioculations na bumubuo ng isang partikular na "frequency rhythm". Ang bilis ng signal ng impulse na dumadaan sa mga peripheral motor fiber ay normal o nababawasan dahil sa pangalawang denervation disorder. [ 10 ]
Ang instrumental diagnosis ay kadalasang kinakatawan din ng ultrasound o MRI ng musculature, na nagpapahintulot sa pagtuklas ng pagpapalit ng kalamnan sa pamamagitan ng mataba na tisyu. Ang MRI ay nagpapakita ng isang tipikal na pattern ng proseso ng pathologic na natatangi sa spinal muscular atrophy. Gayunpaman, ito ay posible lamang sa mga huling yugto ng sugat.
Sa kurso ng morphological analysis ng biopsy ng kalamnan sa mga pasyente, ang isang hindi tiyak na larawan sa anyo ng bundle atrophy at pagpapangkat ng mga fibers ng kalamnan ay tinutukoy. Ang napakaraming bilang ng mga apektadong fibers ng kalamnan ay nabibilang sa uri 1, ang mga katangian ng immunohistological at kemikal ay nasa loob ng normal na mga limitasyon. Ang ultrastructural na larawan ay hindi tiyak.
Ang pinakamahalagang diagnostic procedure para sa pinaghihinalaang spinal muscular atrophy ay ang pagsubok na maaaring makakita ng SMN gene mutation. Sa pamamagitan ng direktang pagsusuri ng DNA, posibleng makita ang presensya o kawalan ng ikapito at ikawalong exon ng SMNc at SMNt genes. Ang pinaka-kaalaman na paraan ay quantitative analysis, na maaaring matukoy ang gene copy number at ipaliwanag ang anyo ng spinal muscular atrophy. Mahalaga rin ang quantitative method sa pagtatasa ng status ng pasyente. Ito ay isang kinakailangang hakbang na isinasagawa para sa layunin ng karagdagang medikal at genetic na pagpapayo sa pamilya.
Ang mga karagdagang diagnostic na pagsusuri ay ginagawa lamang pagkatapos makuha ang negatibong resulta ng pagtanggal ng gene ng SMN. Kung kinakailangan ang pagtuklas ng mga point mutations, maaaring gamitin ang direktang automated sequencing ng SMNT gene.
Iba't ibang diagnosis
Ginagawa ang differential diagnosis sa mga proseso ng pathologic na nagpapakita ng sintomas na kumplikado ng "matamlay na pasyente", na may congenital muscular dystrophies, structural o mitochondrial myopathy. Sa partikular, ang pagkakaroon ng naturang mga pathologies ay dapat na hindi kasama:
- Sakit sa motor neuron;
- Pangunahing lateral myosclerosis;
- Muscular dystrophy;
- Congenital myopathies;
- Mga sakit na nauugnay sa akumulasyon ng glycogen;
- Polio;
- Autoimmune myasthenia gravis.
Ang diagnostic algorithm ay binuo depende sa mga peculiarities ng symptomatology sa isang partikular na bata. Kaya, ang isang espesyal na pag-uuri ng mga pasyente ay ginagamit, depende sa katayuan ng pagganap (Europrotocol TREAT-NMD):
- Hindi makaupo nang walang suporta (nakahiga).
- Nakaupo ngunit hindi makalakad (sedentary).
- May kakayahang kumilos nang nakapag-iisa (mga pasyenteng naglalakad).
Ang sumusunod na diagnostic algorithm ay inirerekomenda para sa mga pasyente sa unang grupo:
- Pisikal na pagsusuri (pagtuklas ng kurbada ng dibdib, pagtatasa ng paggana ng paghinga at pag-ubo, at kondisyon ng balat);
- Pagsubaybay sa puso at paghinga, polysomnography, at pagkakakilanlan ng mga sintomas ng kakulangan sa bentilasyon ng baga;
- Pulse oximetry upang matukoy ang antas ng oxygenation;
- Pagtatasa ng dalas ng mga nakakahawang-namumula na mga pathology at mga kurso ng antibyotiko sa panahon ng matinding anim na buwan;
- Mga x-ray sa dibdib na may paulit-ulit na pag-aaral ng dinamika;
- Pagtatasa ng function ng paglunok.
Para sa mga pasyente sa pangalawang pangkat, nalalapat ang sumusunod na algorithm:
- Pisikal na pagsusulit;
- Pagsubaybay sa puso at paghinga, polysomnography upang makita ang kakulangan sa bentilasyon ng baga;
- Pulse oximetry;
- Pagtatasa ng dalas ng mga nakakahawang proseso ng pamamaga at mga kursong antibyotiko sa matinding anim na buwang panahon;
- Pagsusuri ng spinal column, X-ray ng gulugod, pagtatasa ng antas ng kurbada.
Ang mga pasyente sa ikatlong pangkat ay ipinahiwatig para sa mga naturang pag-aaral:
- Pisikal na pagsusulit;
- Pagsubok sa pag-andar ng paghinga (kabilang ang spirometry, pagkalkula ng dami ng baga, pagtatasa ng function ng kalamnan sa paghinga);
- Upang malaman ang dalas ng mga nakakahawang-namumula na mga pathology at mga kurso sa antibyotiko sa panahon ng matinding taunang panahon.
Ang pagsasagawa ng differential diagnosis ay maaaring kumplikado sa pamamagitan ng pagkakatulad ng SMN1 at SMN2 genes. Upang maiwasan ang mga error, inirerekumenda na gamitin ang paraan ng MLPA, na nagbibigay-daan upang makita ang numero ng kopya ng exon 7 sa SMN1 gene.
Sa karamihan ng mga kaso ng spinal muscular atrophy, mayroong homozygous na pagtanggal ng exon 7 at/o 8 sa SMN1 gene. Gayunpaman, ang ibang mga gene (ATP7A, DCTN1, UBA1, BSCL2, EXOSC3, GARS, atbp.) ay maaari ding maging "mga salarin", na dapat bigyang pansin kung negatibo ang pagsusuri sa SMN1.
Ang biomaterial para sa pag-aaral ay maaaring peripheral blood o fetal blood, dry blood spot map. Ang diagnosis ay sapilitan:
- Sa pagkakaroon ng isang pinalubha na kasaysayan ng spinal muscular atrophy;
- Kapag may nakitang mga kahina-hinalang sintomas, anuman ang namamana na kasaysayan.
Bilang karagdagan, inirerekomenda din ang pananaliksik para sa lahat ng mag-asawa na responsable sa pagpaplano ng pagbubuntis.
Sino ang dapat makipag-ugnay?
Paggamot ng spinal muscular atrophy
Ang mga pasyente na may spinal muscular atrophy ay nangangailangan ng komprehensibong paggamot na kinabibilangan ng:
- Pag-aalaga, tulong, suporta;
- Pagkain sa diyeta;
- Therapy sa droga;
- Mga hakbang sa rehabilitasyon na hindi gamot, kabilang ang kinesiotherapy at physiotherapy.
Ang isang therapeutic regimen na nagsasangkot ng polymodal effect sa lahat ng sistema ng katawan, hindi lamang sa musculoskeletal system, ay pamantayan.
Sa kasamaang palad, imposibleng radikal na pagalingin ang pagkasayang ng kalamnan ng gulugod. Ngunit kadalasan ay posible na mapabuti ang kalidad ng buhay ng pasyente sa pamamagitan ng karampatang paggamit ng mga amino acid at multivitamin complex, neurotrophic agent, calcium channel blockers, vasodilators, cardiotrophic at cytostatic na gamot, protease inhibitors, steroidal drugs, antioxidants, immunoglobulins at immunosuppressants, at iba pa. Napatunayan sa eksperimento na ang paggamot na may mga stem cell, neuroprotective compound at mga molekulang nagpapalakas ng kalamnan ay maaaring humantong sa hindi mahuhulaan na mga systemic disorder. Kasabay nito, ang positibong dinamika pagkatapos ng aplikasyon ng naturang paggamot ay hindi pa napatunayan sa ngayon.
Dahil ang problema ay sanhi ng kakulangan ng normal na protina ng SMN, ang mga pasyente ay maaaring mapabuti sa pamamagitan ng pagtaas ng mga antas ng protina ng SMN ng 25% o higit pa. Para sa kadahilanang ito, ang mga gamot na maaaring mag-activate ng produksyon ng protina na ito ay aktibong sinasaliksik, kabilang ang Gabapentin, Riluzole, Hydroxyurea, Albuterol, valproic acid at sodium phenylbutyrate.
Nag-aalok din ang modernong gamot ng surgical treatment para sa spinal muscular atrophy. Binubuo ito ng surgical alignment ng spinal column - pagwawasto ng neuromuscular curvature. Ang mga surgeon ay nagsasagawa ng multilevel fixation ng gulugod, gamit ang mga espesyal na constructions. Ang sacrum, pelvis, at vertebrae ng upper thoracic o iba pang vertebrae ay ginagamit bilang mga punto ng suporta. Ang operasyon ay nakakatulong upang ihanay ang spinal column, pantay na ipamahagi ang pagkarga dito, alisin ang kakulangan sa ginhawa kapag binabago ang posisyon ng katawan, maiwasan ang masamang epekto sa mga panloob na organo (kabilang ang mga baga).
Mga gamot
Sa kasalukuyan, walang etiologic na paggamot para sa spinal muscular atrophy: ang siyentipikong gamot ay patuloy na gumagana sa gawaing ito. Noong nakaraan, ang mga siyentipiko ay nagtagumpay na sa paghihiwalay ng mga gamot na maaaring mapahusay ang produksyon ng mRNA mula sa SMN2 gene. Ngunit ang malakihang internasyonal na mga klinikal na pagsubok na kinasasangkutan ng mga taong may spinal muscular atrophy ay hindi pa isinasagawa.
Karamihan sa mga gamot na kasama sa karaniwang regimen ng paggamot ay may pangkalahatang prinsipyo ng pagkilos na may medyo mababang katibayan ng pagiging epektibo.
L-Carnitine |
Isang natural na nagaganap na amino acid, isang "kamag-anak" ng mga bitamina ng B-group. Ito ay ginawa sa katawan, naroroon sa atay at nakahalang striated na mga kalamnan, ay kabilang sa isang bilang ng mga sangkap na tulad ng bitamina. Nakikibahagi sa mga proseso ng metabolic, sumusuporta sa aktibidad ng CoA, ay ginagamit upang gawing normal ang metabolismo. Mayroon itong anabolic, antithyroid, antihypoxic na kakayahan, pinasisigla ang metabolismo ng lipid at pag-aayos ng tissue, na-optimize ang gana. Ang L-Carnitine ay inireseta sa halagang humigit-kumulang 1 libong mg bawat araw. Ang kurso ng paggamot ay maaaring tumagal ng hanggang 2 buwan. |
Isang pangkat ng coenzyme benzoquinone na naglalaman ng ilang pangkat ng isoprenyl. Ito ay mga fat-soluble coenzymes, pangunahin na naroroon sa mitochondria ng eukaryotic cellular structures. Ang Ubiquinone ay kasama sa electron transport chain, nakikilahok sa oxidative phosphorylation. Ang pinakamalaking presensya ng sangkap ay matatagpuan sa mga organo na mayaman sa enerhiya - sa partikular, sa atay at puso. Sa iba pang mga bagay, ang coenzyme Q10 ay may mga katangian ng antioxidant, maaaring ibalik ang kapasidad ng antioxidant ng alpha-tocopherol. Karaniwang inireseta mula 30 hanggang 90 mg ng gamot bawat araw, isang dalawang buwang kurso. |
Cerebrolysin |
Isang nootropic na gamot na may mga katangian ng neurotrophic. Madalas itong ginagamit sa mga therapeutic regimen para sa paggamot ng mga neurological pathologies, kabilang ang vascular dementia, stroke. Ang aktibong bahagi ay kinabibilangan ng mga peptide na may limitasyong molekular na timbang na 10 libong dalton. Ang gamot ay ibinibigay bilang isang intravenous injection ng 1-2 ml. Ang kurso ng paggamot ay binubuo ng 10-15 iniksyon. |
Actovegin |
Ang komposisyon ng gamot ay kinakatawan ng mababang molekular na timbang peptides at mga derivatives ng amino acid. Ang Actovegin ay isang hemoderivative: ito ay nakahiwalay sa pamamagitan ng dialysis na may ultrafiltration. Salamat sa paggamit ng gamot, ang pagsipsip at paggamit ng oxygen ay nadagdagan, ang metabolismo ng enerhiya ay pinabilis. Ang gamot ay ginagamit sa anyo ng mga intravenous injection na 1-2 ml, ang kurso ay nangangailangan ng 10-15 injection. |
Solcoseryl |
Ito ay isang deproteinized hemodialysate na may kakayahang mag-optimize ng pre-cellular oxygen at glucose transport, pagpapahusay ng intracellular ATP production, pagpapasigla ng regenerative tissue reactions, pag-activate ng fibroblast proliferation at collagen production sa vascular walls. Ang kurso ng paggamot ay binubuo ng 10-15 intra-muscular injection ng gamot (1-2 ml araw-araw). |
Neuromultivit (bitamina B complex) |
Multivitamin, aktibong ginagamit sa kakulangan ng bitamina B-group. Ito ay madalas na maaaring maging isang kalidad na kapalit para sa isang kurso ng mga iniksyon ng mga paghahanda ng bitamina. I-activate ang mga metabolic na proseso sa utak, nagtataguyod ng pagpapanumbalik ng mga tisyu ng nervous system, ay may analgesic effect. Ang Neuromultivit ay umiinom ng 1-2 tablet araw-araw, isang kurso ng 4 o 8 na linggo. |
Bitamina E |
Isang kilalang antioxidant, bitamina na natutunaw sa taba. Ito ay inireseta sa mga kurso ng 1-2 buwan sa halagang 10-20 IU araw-araw. |
Valproate |
Mayroon silang sedative at relaxing na aktibidad, nagpapakita ng kakayahan sa anticonvulsant, pataasin ang antas ng GABA sa CNS. Ginagamit lamang para sa paggamot ng mga bata na higit sa isang taong gulang, 10 hanggang 20 mg bawat kg bawat araw. |
Salbutamol |
Isang bronchodilator, na kabilang sa pangkat ng mga pumipili na beta2-adrenoreceptor agonist. Ang regular na paggamit ng gamot ay nagdudulot ng pagtaas ng produksyon ng mRNA at SMN protein, na positibong nakakaapekto sa klinikal na larawan ng spinal muscular atrophy. Ang salbutamol ay ginagamit nang maingat, 2-4 mg apat na beses sa isang araw (ang maximum na halaga ay 32 mg bawat araw). |
Ang isa sa mga pinakabagong gamot na ginagamit sa spinal muscular atrophy ay ang Zolgensma genotherapeutic na gamot na Zolgensma, na nagsisiguro sa aktibidad at tamang paggana ng transduced motor nerve cells. Ang gamot ay pinangangasiwaan kasama ng mga immunomodulatory na gamot ayon sa isang espesyal na protocol at pinangangasiwaan nang isang beses sa intravenously, batay sa isang nominal na dosis na 1.1 ͯ 1014 Vg/kg (ang kabuuang dami ng pangangasiwa ay tinutukoy depende sa bigat ng pasyente).
Bago simulan ang paggamot sa Zolgensma, ipinag-uutos na matukoy ang antas ng mga antibodies sa AAV9 gamit ang isang napatunayang pamamaraan ng diagnostic, tasahin ang hepatic function (ALT, AST, kabuuang bilirubin), magsagawa ng pangkalahatang pagsusuri sa klinikal na dugo at pagsusuri ng troponin I, matukoy ang antas ng creatinine. Kung ang talamak at talamak na aktibong nakakahawang kondisyon ay nakita, ang pangangasiwa ng gamot ay ipinagpaliban hanggang sa gumaling o makumpleto ang yugto ng pagbabalik ng nakakahawang proseso.
Ang pinaka-madalas na side effect ng gamot ay itinuturing na pagkabigo sa atay, na maaaring nakamamatay.
Iba pang mga aprubadong gamot na maaaring ireseta ng iyong doktor para sa pagkasayang ng kalamnan ng gulugod:
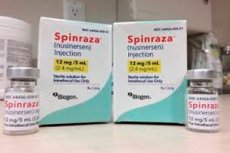
- Ang Spinraza ay isang paghahanda ng nusinersen sodium, isang antisense oligonucleotide na partikular na idinisenyo para sa paggamot ng spinal amyotrophy. Ito ay inilaan para sa intrathecal na pangangasiwa sa pamamagitan ng lumbar puncture. Ang inirekumendang dosis ay 12 mg. Ang regimen ng paggamot ay tinutukoy ng dumadating na manggagamot.
- Ang Risdiplam ay isang gamot na binabago ang splicing ng mRNA precursor ng motor nerve cell survival gene 2. Ang Risdiplam ay iniinom nang pasalita, isang beses sa isang araw. Ang dosis ay tinutukoy ng doktor nang paisa-isa, isinasaalang-alang ang edad at bigat ng pasyente. Ang paggamit ng gamot sa mga batang wala pang 2 buwan ay kontraindikado. Ang embryofetal toxicity ng gamot na ito ay nabanggit, kaya ang reproductive-potential na mga pasyente ay dapat magsagawa ng maingat na contraceptive measures sa panahon at ilang panahon pagkatapos ng paggamot.
Physiotherapeutic na paggamot para sa spinal muscular atrophy
Ang Physiotherapy ay ginagamit bilang isa sa mga link ng kumplikadong therapy at rehabilitasyon ng mga pasyente na may spinal muscular atrophy. Ang mga pangunahing punto ng naturang paggamot ay:
- Paggamit ng pagbabawas sa pamamagitan ng mga sistema ng suspensyon, aktibong-passive na pagsasanay, paggamit ng percutaneous electrical stimulation ng spinal cord;
- Mga ehersisyo sa paghinga at pisikal na therapy;
- Kalahating oras na verticalization session;
- Translingual electrostimulation treatment (20-minutong session, na sinamahan ng mga ehersisyo upang mapabuti ang mahusay na mga kasanayan sa motor);
- Mga manu-manong pamamaraan;
- Mga aplikasyon ng paraffin sa iba't ibang grupo ng mga artikulasyon;
- Darsonval upang mapabuti ang pagganap ng kalamnan.
Ang paraan ng darsonvalization ay batay sa epekto sa mga tisyu gamit ang isang alternating high-frequency pulse current ng mataas na boltahe at mababang lakas. Pagkatapos ng isang kurso ng mga pamamaraan, mayroong isang pagtaas sa pagganap ng kalamnan, pagpapalakas ng microcirculation, pagpapalawak ng mga arterioles at capillaries, pag-aalis ng ischemia, pagpapabuti ng nutrisyon at supply ng oxygen sa mga kalamnan, na may positibong epekto sa kurso ng mga proseso ng pagbabagong-buhay at atrophic.
Ang isa sa mga pinaka makabuluhang problema sa mga pasyente na may spinal amyotrophy ay ang panghihina ng kalamnan sa paghinga, na kadalasang humahantong sa respiratory dysfunction at pagkamatay ng pasyente.
Sa spinal amyotrophy, ang buong skeletal musculature, kabilang ang responsable para sa paghinga, ay hindi gumaganap. Ang kahinaan at unti-unting pagkasayang ng kalamnan ay nakakaapekto sa kalidad ng respiratory act, humahantong sa pag-unlad ng mga komplikasyon at pagtaas ng respiratory failure. Samakatuwid, kinakailangan na gumawa ng mga hakbang upang palakasin ang mga kalamnan, ang pag-iwas sa mga komplikasyon sa paghinga at mga impeksyon sa respiratory tract. Ang isang espesyal na papel sa ito ay gumaganap ng gymnastics na may Ambu bag, na isinasagawa kasabay ng physical therapy, stretching exercises, massage. Ang paggamit ng Ambu bag ay nagpapahintulot sa iyo na "palawakin" ang dami ng dibdib at baga. Para sa mga aktibidad ng mga bata ay angkop na bag na may dami ng hindi bababa sa isa at kalahating litro, nilagyan ng balbula upang palabasin ang labis na presyon (upang maiwasan ang barotrauma).
Ang mga ehersisyo ay hindi dapat gawin sa isang buong tiyan. Posisyon ng katawan - nakaupo, semi-upo, nakahiga sa gilid o likod (kung walang mga problema sa plema): pinakamainam na gawin ang mga pamamaraan sa iba't ibang posisyon sa bawat oras. Mahalaga na ituwid ang likod ng pasyente. Kung kinakailangan, ginagamit ang isang korset. Bago simulan ang pamamaraan, siguraduhin na ang daanan ng hangin ay walang plema.
Masahe para sa spinal muscular atrophy
Ang masahe para sa paggamot ng spinal amyotrophy ay dapat na magaan at banayad. Sa mga lugar na may maskuladong resistensya ay nalalapat ang mga pangkalahatang epekto, kabilang ang pag-tap, at sa mga lugar na napanatili ang innervation ay gumamit ng malalim na paghaplos (paayon, nakahalang), pagmamasa.
Sa pangkalahatan, ang pagsasanay ng iba't ibang uri ng masahe, depende sa mga indibidwal na katangian ng kurso ng sakit, ang edad ng pasyente. Ang mga ito ay maaaring:
- Pagmamasa upang pasiglahin ang malalim na mga kalamnan;
- Kuskusin upang ma-optimize ang sirkulasyon ng dugo at lymph;
- Spot treatment ng mga trigger point;
- Ng fiber-strengthening pounding.
Mahalaga na ang epekto ay kumalat sa buong lugar ng problema.
Contraindications sa masahe para sa spinal muscular atrophy:
- Talamak na pamamaga, mataas na temperatura ng katawan;
- Mga karamdaman sa dugo, mga tendensya sa pagdurugo;
- Mga proseso ng purulent;
- Nakakahawa, fungal dermatologic na sakit;
- Vascular aneurysms, thrombangiitis, endarteritis, lymphadenitis;
- Benign at malignant neoplasms.
Ang kurso ng anumang masahe para sa isang pasyente na may spinal muscle atrophy ay mahigpit na inireseta nang paisa-isa. Ang hindi wastong pagsasagawa ng pamamaraan, ang labis na magaspang at hindi tamang epekto ay maaaring makapinsala sa kondisyon ng pasyente.
Pag-iwas
Ang direkta at hindi direktang pagsusuri sa DNA at pagsusuri sa prenatal na DNA ay aktibong ginagawa na ngayon. Ito ay makabuluhang binabawasan ang posibilidad ng isang may sakit na sanggol na maipanganak, na lalong mahalaga para sa mga mag-asawa na nakaranas na ng kapanganakan ng mga bata na may spinal muscular atrophy.
Ang mga hakbang sa pag-iwas ay kumakatawan sa isang mahalagang medikal na takbo at ikinategorya sa pangunahin, pangalawa at pangatlong hakbang.
Ang mga pangunahing hakbang ay naglalayong direktang pigilan ang impluwensya ng isang hindi kanais-nais na kadahilanan at maiwasan ang pag-unlad ng sakit. Ang ganitong pag-iwas ay binubuo sa pagwawasto ng diyeta at pang-araw-araw na pamumuhay, na humahantong sa isang malusog na pamumuhay.
Ang pangalawang pag-iwas ay binubuo sa pag-aalis ng mga halatang kadahilanan ng panganib at kasama ang maagang pagsusuri ng mga pathology, pagtatatag ng pagsubaybay sa dinamika, nakadirekta na paggamot.
Ang pag-iwas sa tertiary ay isinasagawa kaugnay sa isang taong may sakit na pinagkaitan ng ilang mga kakayahan sa motor. Sa sitwasyong ito, pinag-uusapan natin ang tungkol sa gamot, sikolohikal, panlipunan at rehabilitasyon sa paggawa.
Ayon sa impormasyon mula sa World Health Organization, higit sa 2% ng mga sanggol sa mundo ay ipinanganak na may ilang uri ng developmental disorder. Kasabay nito, 0.5-1% ng naturang mga karamdaman ay nagmula sa genetic na pinagmulan. Ang pag-iwas sa naturang mga problema ay nabawasan sa medikal na genetic counseling at kalidad ng prenatal diagnosis, na nagbibigay-daan sa pagliit ng mga panganib ng panganganak ng isang sanggol na may genetic pathology.
Ang panganib ng isang tao na magkaroon ng spinal muscular atrophy o ibang genetic disease ay depende sa mga gene na minana mula sa kanyang ina at ama. Ang maagang pagkakakilanlan ng namamana na mga kadahilanan, pagkalkula ng mga indibidwal na panganib ng genetically tinutukoy na patolohiya ay maaaring tinatawag na isang paraan ng naka-target na pag-iwas.
Kasama sa mga hakbang sa diagnostic ng prenatal ang direkta at hindi direktang pamamaraan ng pananaliksik. Sa una, ang mga kababaihan na nangangailangan ng hindi direktang prenatal diagnosis ay natukoy. Maaaring kabilang dito ang:
- Mga buntis na kababaihan 35 taong gulang at mas matanda;
- Na nagkaroon ng 2 o higit pang nakaraang kusang pagpapalaglag;
- Sino ang may mga anak na may genetic developmental defects;
- Na may hindi kanais-nais na namamana na kasaysayan;
- Na nagkaroon ng mga impeksyon sa viral o pagkakalantad sa radiation (kabilang sa yugto ng pagpaplano ng pagbubuntis).
Para sa mga layuning pang-iwas, ang mga pamamaraan tulad ng ultrasound, hormonal test (biochemical screening) ay ginagamit. Minsan ginagamit din ang mga invasive procedure tulad ng chorionbiopsy, amniocentesis, placentocentesis, cordocentesis. Ang mapagkakatiwalaang impormasyon tungkol sa mga genetic na panganib ay nagbibigay-daan sa iyo na ayusin ang iyong pamumuhay at pagbubuntis upang maiwasan ang pagsilang ng isang maysakit na bata.
Bakuna sa spinal muscular atrophy
Siyempre, ang lahat ng mga magulang ng mga bata na may spinal amyotrophy ay nais na ganap na pagalingin sila ng sakit. Gayunpaman, walang bakuna na maaaring matanggal ang problema. Kahit na ang pananaliksik sa pag-optimize ng paggamot ay patuloy.
Sa partikular, noong 2016, inaprubahan ng mga Amerikanong siyentipiko ang natatanging gamot na Spinraza (nusinersen), na kasunod na inaprubahan para gamitin sa mga bansang Europeo.
Sinisiyasat ng mga espesyalista ang problema ng paggamot sa spinal muscular atrophy sa mga ganitong paraan:
- Pag-aayos o pagpapalit ng "maling" SMN1 gene;
- Potentiation ng function ng normal na SMN2 gene;
- Proteksyon ng motor nerve cells na apektado dahil sa kakulangan sa protina ng SMN;
- Proteksyon ng mga kalamnan mula sa mga pagbabago sa atrophic upang maiwasan o maibalik ang nawalang pag-andar laban sa background ng pag-unlad ng patolohiya.
Ang therapy sa gene ay nagsasangkot ng pag-target sa nasirang gene gamit ang mga viral vector na dumadaan sa lamad ng dugo-utak at umaabot sa naaangkop na lugar sa spinal cord. Pagkatapos, ang virus ay "nakakahawa" sa apektadong selula na may malusog na bahagi ng DNA, na parang "nagsusuri" sa depekto ng gene. Kaya, ang pag-andar ng mga cell nerve ng motor ay naitama.
Ang isa pang direksyon ay maliit na molecule therapy, ang kakanyahan nito ay upang mapahusay ang pag-andar ng SMN2 gene. Ang mga sanggol na may diagnosed na spinal muscular atrophy ay mayroong kahit isang kopya ng SMN2 gene. Ang direksyong ito ay aktibong sinaliksik ng mga Amerikanong siyentipiko, at sa ngayon, maraming mga gamot na naglalayong pahusayin ang synthesis ng isang kumpletong protina mula sa gene ng SMN2 ay sumasailalim sa mga klinikal na pagsubok.
Ang isa pang paraan ng posibleng therapeutic intervention ay upang galugarin ang neuroprotection upang mabawasan ang pagkamatay ng motor neuron, dagdagan ang kanilang kakayahang umangkop at pagbutihin ang pag-andar.
Ang ikatlong direksyon ay nagsasangkot ng pagprotekta sa kalamnan mula sa mga proseso ng atrophic. Dahil ang kakulangan sa protina ng SMN ay negatibong nakakaapekto sa mga cell nerve ng motor at function ng kalamnan, ang layunin ng paggamot na ito ay dapat na protektahan ang mga kalamnan mula sa pagkasayang, dagdagan ang mass ng kalamnan, at ibalik ang function ng kalamnan. Ang ganitong uri ng therapy ay hindi makakaapekto sa genetic apparatus, ngunit maaari itong bumagal o kahit na hadlangan ang paglala ng spinal muscular atrophy.
Screening para sa spinal muscular atrophy
Ang bagong panganak na screening ay lalong ginagamit sa medikal na kasanayan at kadalasan ay gumaganap ng isang mapagpasyang papel. Ang pagtuklas ng spinal muscular atrophy sa lalong madaling panahon ay maaaring makabuluhang mapabuti ang pagbabala para sa may sakit na bata. Kasama sa pagsusuri sa pagsusuri ang mga sumusunod na puntos na nakabalangkas sa talahanayan:
Isang anyo ng spinal muscular atrophy |
Symptomatology |
Spinal muscular atrophy type I (ang bata ay hindi maaaring umupo, average na pag-asa sa buhay - hanggang 2 taon) |
Ito ay nagpapakita ng sarili mula sa kapanganakan hanggang sa edad na anim na buwan. Ang hindi sapat na tono ng kalamnan ay nakita, ang pag-iyak ay mahina, ang kahinaan ng kalamnan (kabilang ang pagnguya at paglunok ng mga kalamnan) ay tumataas. May mga problema sa pagpapanatili ng ulo, ipinapalagay ng sanggol ang isang "palaka" na postura kapag nakahiga. |
Spinal muscular atrophy type II (ang bata ay nakakaupo, ang pag-asa sa buhay ay karaniwang higit sa 2 taon, at higit sa kalahati ng mga pasyente ay nabubuhay hanggang 20-25 taong gulang) |
Nagde-debut ito simula sa edad na 7 buwan at hanggang isa at kalahating taong gulang. Minsan napapansin ang mga problema sa paglunok, paghinga at pag-ubo. Ang mga permanenteng palatandaan ay kinabibilangan ng muscle spasms, limitadong joint mobility, curvature ng spinal column, mababang presyon ng dugo at panghina ng kalamnan. |
Spinal muscular atrophy type III (ang bata ay maaaring umupo at lumipat, ngunit ang mga kakayahan sa itaas ay unti-unting nawawala, ang pag-asa sa buhay ay normal) |
Mga debut sa edad na isa at kalahating taon. Ang kurbada ng spinal column at thorax, muscular atrophy ng pelvis at proximal legs, at pagtaas ng joint mobility ay nabanggit. Mahirap ang paglunok. |
Uri ng spinal muscular atrophy IV |
Tumutukoy sa pormang pang-adulto. Ang Symptomatology ay magkapareho sa uri ng spinal muscular atrophy na III. Unti-unting tumataas ang kahinaan, lumilitaw ang mga panginginig at fascioculations ng kalamnan sa debut sa edad na 16-25. |
Pagtataya
Sa Werdnig-Hoffman syndrome, ang average na pag-asa sa buhay ay 1.5-2 taon. Ang nakamamatay na kinalabasan sa karamihan ng mga kaso ay dahil sa pagtaas ng respiratory failure at pag-unlad ng pamamaga sa mga baga. Sa napapanahong suporta sa paghinga sa anyo ng artipisyal na bentilasyon, posible na bahagyang mapataas ang pag-asa sa buhay ng sanggol. Mayroong espesyal na pangangailangan para sa tuluy-tuloy na palliative na pangangalaga, na kinakailangan din sa spinal amyotrophy type II. Ang mga pathology ng ikatlo at ikaapat na uri ay nailalarawan sa pamamagitan ng isang mas kanais-nais na pagbabala.
Ang anumang uri ng spinal muscular atrophy ay isang malubhang sakit. Ang lahat ng miyembro ng pamilya ng pasyente ay nangangailangan ng patuloy na sikolohikal, impormasyon at panlipunang suporta. Mahalaga para sa pasyente na matiyak ang sapat na diagnosis at propesyonal na suporta mula sa mga espesyalista tulad ng pediatrician, neurologist, neurologist, pulmonologist, cardiologist, orthopedist, physiotherapist, atbp. Sa kabila ng kakulangan ng partikular na therapy para sa sakit, ang symptomatic na paggamot ay isinasagawa, ang espesyal na nutrisyon ay inireseta (parehong parenteral at enteral), iba't ibang mga hakbang sa rehabilitasyon at pag-iwas sa pag-unlad ng rehabilitasyon. mga komplikasyon.
Maraming mga pasyente ang binibigyan ng kapansanan, at ang isang indibidwal na pamamaraan ng rehabilitasyon ay iginuhit.
Ang natural na nagaganap na spinal muscular atrophy nang walang paggamit ng mga espesyal na kagamitan upang suportahan ang paghinga at pagpapakain sa halos kalahati ng mga kaso ay nagtatapos sa pagkamatay ng maysakit na bata bago ang edad ng dalawang taon (karamihan ay type I disease).