Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Usher syndrome
Huling nasuri: 04.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Ang Usher syndrome ay isang namamana na sakit na nagpapakita ng sarili bilang kumpletong pagkabingi mula sa kapanganakan, pati na rin ang progresibong pagkabulag sa edad. Ang pagkawala ng paningin ay nauugnay sa retinitis pigmentosa, isang proseso ng pigmentary degeneration ng retina. Maraming tao na may Usher syndrome ay mayroon ding malubhang problema sa balanse.
Epidemiology
Salamat sa pananaliksik, posible na maitatag na ang Usher syndrome ay nakakaapekto sa halos 8% ng mga nasuri na bingi-mute na mga bata (ang mga pagsusulit ay isinagawa sa mga espesyal na institusyon para sa mga taong bingi-mute). Ang pigmentary retinitis ay sinusunod sa 6-10% ng mga pasyente na dumaranas ng congenital deafness, na kung saan, ay sinusunod sa halos 30% ng mga taong may pigmentary retina disease.
Ito ay pinaniniwalaan na ang sakit na ito ay nagpapakita mismo sa humigit-kumulang 3-10 katao sa 100 libo sa buong mundo. Maaari itong maobserbahan sa parehong babae at lalaki nang pantay. Humigit-kumulang 5-6% ng populasyon ng mundo ang nagdurusa sa sindrom na ito. Humigit-kumulang 10% ng lahat ng kaso ng childhood profound deafness ay nangyayari dahil sa Usher syndrome I, pati na rin sa mga uri ng II.
Sa Estados Unidos, ang mga uri 1 at 2 ay ang pinakakaraniwang mga uri. Magkasama, ang mga ito ay nagkakahalaga ng humigit-kumulang 90 hanggang 95 porsiyento ng lahat ng kaso ng Usher syndrome sa mga bata.
Mga sanhi Usher syndrome
Ang mga uri ng Usher syndrome na I, II, at III ay may autosomal recessive na sanhi, habang ang uri IV ay itinuturing na isang X-chromosome disorder. Ang mga sanhi ng pagkabulag at pagkabingi na nangyayari sa sindrom na ito ay hindi pa sapat na pinag-aralan. Ipinapalagay na ang mga taong may sakit na ito ay sobrang sensitibo sa mga sangkap na maaaring makapinsala sa istraktura ng DNA. Bilang karagdagan, ang sakit na ito ay maaaring nauugnay sa mga karamdaman sa immune system, ngunit sa kasong ito, walang eksaktong larawan ng prosesong ito.
Noong 1989, unang natukoy ang mga abnormalidad ng chromosomal sa mga pasyenteng may type II na sakit, na sa hinaharap ay maaaring humantong sa isang paraan upang ihiwalay ang mga gene na nagdudulot ng sindrom. Posible ring matukoy ang mga gene na ito sa mga carrier at bumuo ng mga espesyal na prenatal genetic na pagsusuri.
 [ 8 ]
[ 8 ]
Mga kadahilanan ng peligro
Ang sindrom ay minana kapag ang parehong mga magulang ay apektado, ibig sabihin, ito ay minana ng isang recessive na uri. Ang isang bata ay maaari ring magmana ng sakit kung ang kanyang mga magulang ay carrier ng gene. Kung ang parehong mga hinaharap na magulang ay may ganitong gene, kung gayon ang posibilidad na magkaroon ng isang sanggol na may ganitong sindrom ay 1 sa 4. Ang isang tao na mayroon lamang isang gene para sa sindrom ay itinuturing na isang carrier, ngunit walang mga sintomas ng disorder. Sa ngayon, hindi pa matukoy kung ang isang tao ay may gene para sa sakit na ito.
Kung ang isang bata ay ipinanganak sa mga magulang, ang isa sa kanila ay walang ganoong gene, kung gayon ang posibilidad na siya ay magmana ng sindrom ay napakababa, ngunit tiyak na siya ay magiging isang carrier.
Mga sintomas Usher syndrome
Kasama sa mga sintomas ng Usher syndrome ang pagkawala ng pandinig at abnormal na akumulasyon ng mga pigmented na selula sa mga istruktura ng mata. Ang pasyente ay magkakaroon ng pagkabulok ng retina, na nagiging sanhi ng pagkasira ng paningin at pagkawala ng paningin sa mga pinakamalalang kaso.
Ang pagkawala ng pandinig sa sensorineural ay maaaring banayad o kumpleto at kadalasan ay hindi umuunlad mula sa pagsilang. Gayunpaman, ang retina pigment disease ay maaaring magsimulang umunlad sa pagkabata o mamaya. Ipinakita ng mga resulta ng pagsusuri na ang central visual acuity ay maaaring mapanatili sa loob ng maraming taon, kahit na lumala ang peripheral vision (isang kondisyon na tinatawag na "tunnel vision").
Ito ang mga pangunahing pagpapakita ng sakit, na kung minsan ay maaaring madagdagan ng iba pang mga karamdaman, tulad ng psychosis at iba pang mga sakit sa pag-iisip, mga problema sa panloob na tainga at/o mga katarata.
Mga Form
Sa panahon ng pananaliksik, 3 uri ng sakit na ito ang natukoy, pati na rin ang isang ika-4 na anyo, na medyo bihira.
Ang Type I ng sakit ay nailalarawan sa pamamagitan ng congenital complete deafness, pati na rin ang balance disorder. Kadalasan, ang mga naturang bata ay nagsisimulang maglakad lamang sa edad na 1.5 taon. Ang pagkasira ng paningin ay karaniwang nagsisimula sa edad na 10, at ang huling pag-unlad ng estado ng pagkabulag sa gabi ay nagsisimula sa edad na 20. Ang mga bata na may ganitong uri ng sakit ay maaaring magkaroon ng progresibong pagkasira ng peripheral vision.
Sa type II na sakit, ang katamtaman o congenital na pagkabingi ay sinusunod. Sa kasong ito, ang pagkasira sa bahagyang pagkabingi ay kadalasang hindi na nangyayari. Ang pigmentary retinitis ay nagsisimulang umunlad sa pagtatapos ng pagbibinata o pagkatapos ng 20 taon. Ang pag-unlad ng pagkabulag sa gabi ay karaniwang nagsisimula sa 29-31 taon. Ang kapansanan sa visual acuity sa kaso ng type II na patolohiya ay karaniwang umuunlad nang kaunti nang mas mabagal kaysa sa uri I.
Ang uri III ng sakit ay nailalarawan sa pamamagitan ng progresibong pagkawala ng pandinig, kadalasang nagsisimula sa panahon ng pagdadalaga, pati na rin ang unti-unting pag-unlad sa parehong panahon (medyo mas huli kaysa sa pagkawala ng pandinig) ng retinitis pigmentosa, na maaaring maging isang kadahilanan sa pag-unlad ng progresibong pagkabulag.
Ang mga pagpapakita ng uri ng patolohiya ng IV ay pangunahing nangyayari sa mga lalaki. Sa kasong ito, ang mga progresibong karamdaman at pagkawala ng pandinig at paningin ay sinusunod din. Ang form na ito ay napakabihirang at kadalasan ay may likas na X-chromosomal.
Diagnostics Usher syndrome
Ang diagnosis ng Usher syndrome ay ginawa batay sa naobserbahang kumbinasyon ng pasyente ng biglaang pagkabingi at progresibong pagkawala ng paningin.
Mga pagsubok
Maaaring mag-utos ng isang espesyal na genetic test upang matukoy ang mutation.
Labing-isang genetic loci ang natagpuan na maaaring maging sanhi ng pag-unlad ng Usher syndrome, at siyam na mga gene ang natukoy na tiyak na sanhi ng disorder:
- Uri 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Uri 2: ush2a, VLGR1, WHRN.
- Uri ng Usher syndrome 3: USH3A.
Natukoy ng mga siyentipiko ng NIDCD, kasama ang mga kasamahan mula sa mga unibersidad sa New York at Israel, ang isang mutation na tinatawag na R245X sa Pcdh15 gene na bumubuo ng malaking porsyento ng type 1 Usher syndrome sa populasyon ng mga Hudyo.
Upang malaman ang tungkol sa mga lab na nagsasagawa ng mga klinikal na pagsubok, bisitahin ang https://www.genetests.org at hanapin ang direktoryo ng lab para sa "Usher syndrome."
Upang matutunan ang tungkol sa mga kasalukuyang klinikal na pagsubok na kinabibilangan ng genetic testing para sa Usher syndrome, bisitahin ang https://www.clinicaltrials.gov at hanapin ang "Usher syndrome" o "Usher syndrome genetic testing."
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Mga instrumental na diagnostic
Mayroong ilang mga paraan ng instrumental diagnostics:
- Pagsusuri ng fundus upang makita ang pagkakaroon ng mga pigment spot sa retina, pati na rin ang pagpapaliit ng mga retinal vessel;
- Electroretinogram, na nagbibigay-daan upang makita ang mga paunang degenerative deviations sa retina ng mata. Ipinapakita nito ang pagkalipol ng mga electroradiographic pathways;
- Ang isang electronystagmogram (ENG) ay sumusukat sa mga hindi boluntaryong paggalaw ng mata na maaaring magpahiwatig ng pagkakaroon ng kawalan ng timbang.
- Audiometry, na ginagamit upang matukoy ang pagkakaroon ng pagkabingi at ang kalubhaan nito.
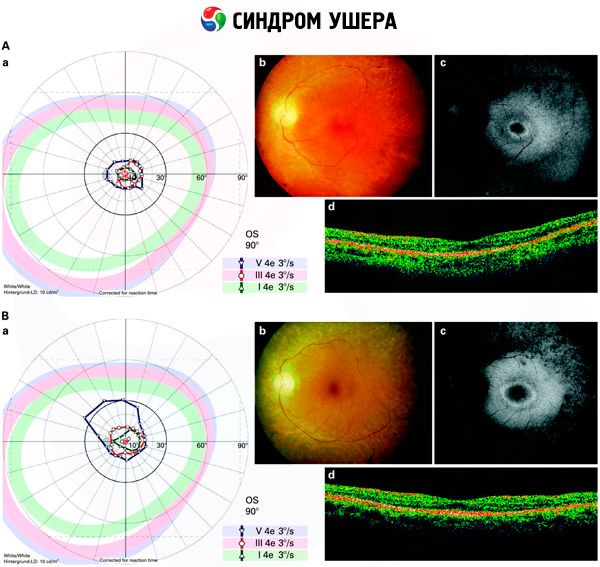
Iba't ibang diagnosis
Ang Usher syndrome ay dapat na naiiba sa ilang katulad na mga karamdaman.
Hallgren syndrome, na kung saan ay nailalarawan sa pamamagitan ng congenital hearing loss at progressive vision loss (cataracts at nystagmus also develop). Kasama sa mga karagdagang sintomas ang ataxia, psychomotor disorder, psychosis, at mental retardation.
Alstrom syndrome, na isang namamana na sakit kung saan ang retina ay bumababa, na nagreresulta sa pagkawala ng gitnang paningin. Ang sindrom na ito ay nauugnay sa labis na katabaan ng pagkabata. Kasabay nito, ang diabetes mellitus at pagkawala ng pandinig ay nagsisimulang umunlad pagkatapos ng 10 taon.
Ang rubella sa isang buntis sa unang trimester ay maaaring magdulot ng iba't ibang abnormalidad sa pag-unlad ng bata. Kabilang sa mga kahihinatnan ng naturang abnormalidad ay ang pagkawala ng pandinig, gayundin ang (o) mga problema sa paningin, at bilang karagdagan dito, iba't ibang mga depekto sa pag-unlad.
Sino ang dapat makipag-ugnay?
Paggamot Usher syndrome
Sa kasalukuyan ay walang lunas para sa Usher syndrome. Samakatuwid, ang therapy sa kasong ito ay higit sa lahat ay binubuo ng pagbagal sa proseso ng pagkawala ng paningin, pati na rin ang pagbabayad para sa pagkawala ng pandinig. Ang mga posibleng paraan ng paggamot ay kinabibilangan ng:
- Ang pag-inom ng bitamina A (naniniwala ang ilang mga ophthalmologist na ang mataas na dosis ng bitamina A palmitate ay maaaring makapagpabagal, ngunit hindi huminto, ang pag-unlad ng retinitis pigmentosa);
- Ang pagtatanim ng mga espesyal na elektronikong aparato sa mga tainga ng pasyente (hearing aid, cochlear implants.
Inirerekomenda ng mga ophthalmologist na ang karamihan sa mga nasa hustong gulang na may karaniwang mga anyo ng retinitis pigmentosa ay kumuha ng 15,000 IU (internasyonal na mga yunit) ng bitamina A palmitate araw-araw sa ilalim ng pangangasiwa. Dahil ang mga taong may type 1 Usher syndrome ay hindi kasama sa pag-aaral, ang mataas na dosis ng bitamina A ay hindi inirerekomenda para sa grupong ito ng mga pasyente. Ang mga taong isinasaalang-alang ang pagkuha ng bitamina A ay dapat talakayin ang opsyon sa paggamot na ito sa kanilang doktor. Ang iba pang mga rekomendasyon para sa opsyon sa paggamot na ito ay kinabibilangan ng:
- Pagbabago ng iyong diyeta upang isama ang mga pagkaing mataas sa bitamina A.
- Ang mga babaeng nagpaplanong magbuntis ay dapat huminto sa pag-inom ng mataas na dosis ng bitamina A tatlong buwan bago sila magplanong magbuntis dahil sa mas mataas na panganib ng mga depekto sa kapanganakan.
- Ang mga babaeng buntis ay dapat huminto sa pag-inom ng mataas na dosis ng bitamina A dahil sa mas mataas na panganib ng mga depekto sa kapanganakan.
Mahalaga rin na maiangkop ang naturang bata sa buhay panlipunan. Nangangailangan ito ng tulong ng mga guro at psychologist ng espesyal na edukasyon. Kung ang pasyente ay nagsimulang makaranas ng progresibong pagkawala ng paningin, dapat siyang turuan na gumamit ng sign language.
Pagtataya
Ang Usher syndrome ay may hindi kanais-nais na pagbabala. Ang visual field at ang katalinuhan nito ay nagsisimulang lumala sa panahon ng 20-30 taon sa karamihan ng mga pasyente na may ganitong sakit ng anumang uri. Sa ilang mga kaso, ang kumpletong bilateral na pagkawala ng paningin ay nangyayari. Ang pagkawala ng pandinig, na palaging sinasamahan ng pagkapipi, ay napakabilis na nabubuo upang makumpleto ang bilateral na pagkawala ng pandinig.