Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Angelman syndrome sa mga bata at matatanda
Huling nasuri: 04.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.

Mayroong ilang mga sakit kung saan ang mga expression tulad ng "ingatan ang iyong sarili at hindi ka magkakasakit" ay tunog, sa pinakadulo, katawa-tawa. Ang mga ito ay mga pathology kung saan ang ilang mga mental at pisikal na abnormalidad ay likas sa katawan ng bata bago pa man ipanganak, ngunit ang mga magulang ay hindi dapat sisihin para dito. Ang mga ganitong sakit ay sanhi ng mga mutasyon o abnormalidad sa mga chromosome set at tinatawag na chromosomal o genetic. Angelman syndrome, Down syndrome, Patau syndrome, Edwards syndrome, Turner syndrome, Prader-Willi syndrome - ito ay bahagi lamang ng mga genetic na sakit mula sa isang medyo disenteng listahan.
Happy Man Syndrome
Sa oras na ito ay pag-uusapan natin ang tungkol sa patolohiya na pinangalanan sa Ingles na pediatrician na si Harry Angelman, na unang nagtaas ng isyu ng problemang ito noong 1965, na nakatagpo ng tatlong hindi pangkaraniwang mga bata sa kanyang pagsasanay noong nakaraang araw, na pinagsama ng mga karaniwang kakaibang sintomas. Tinawag ng doktor ang mga batang ito na manika at nagsulat ng isang artikulo tungkol sa kanila, na noong una ay tinawag na "Mga Bata-marionette". Ang artikulo mismo at ang pamagat nito ay isinulat sa ilalim ng impresyon ng isang pagpipinta na nakita sa isa sa mga museo ng Verona. Ang pagpipinta ay naglalarawan ng isang tumatawa na batang lalaki, at tinawag itong "The Puppet Boy". Ang pagkakaugnay ng bata na inilalarawan sa pagpipinta kasama ang tatlong bata na minsang nakatagpo ni Angelman sa kanyang pagsasanay ay nagtulak sa pediatrician na pagsamahin ang mga bata sa isang grupo dahil sa sakit na mayroon sila.
Walang nakakagulat na ang mga batang binanggit sa artikulo ay hindi napansin ng ibang mga doktor. Pagkatapos ng lahat, sa unang sulyap ay tila mayroon silang ganap na magkakaibang mga sakit, kaya iba ang pangkalahatang klinikal na larawan ng sakit sa 3 magkakaibang mga kaso. Marahil ang "bagong" chromosomal pathology ay magkakaroon ng interes sa iba pang mga siyentipiko, ngunit sa oras na iyon ang genetika ay hindi pa binuo upang kumpirmahin ang hypothesis ng Ingles na doktor. Samakatuwid, pagkatapos ng isang tiyak na interes dito, ang artikulo ay itinapon sa likod na istante sa loob ng mahabang panahon.
Ang susunod na pagbanggit ng Angelman syndrome, na kung ano ang tawag ngayon sa artikulo ng Ingles na pediatrician na si G. Angelman, ay nagsimula noong unang bahagi ng 80s ng ika-20 siglo. At noong 1987 lamang posible na mahanap ang dahilan kung bakit ang isang maliit na bahagi ng mga bata ay ipinanganak na may tulad na mga paglihis na mula sa labas ay tila sila ay patuloy na nakangiti at masaya. Sa katunayan, ito ay hindi totoo sa lahat, at ang ngiti ay isang pagngiwi lamang, sa likod nito ay nagtatago ng isang malungkot na kaluluwa ng tao at ang sakit ng mga magulang.
Epidemiology
Ayon sa mga istatistika, ang isang chromosomal mutation sa isang bata ay maaaring bumuo kapwa laban sa background ng mga katulad na mutasyon sa mga magulang at sa kawalan ng ganoon. Walang malinaw na namamana na katangian ng Angelman syndrome (AS), ngunit ang posibilidad na magkaroon ng patolohiya sa mga magulang na may mga mutasyon ng chromosomal ay medyo mataas.
Nakatutuwa rin na kung ang isang pamilya ay mayroon nang anak na may AS, mayroong isang porsyentong posibilidad na magkaroon ng pangalawang anak na may parehong karamdaman, kahit na ang mga magulang ay malusog.
Wala pa ring eksaktong istatistika sa bilang ng mga pasyente na may Angelman syndrome. Marahil ang dahilan ay ang iba't ibang mga sintomas, na maaaring mangyari sa isang tiyak na komposisyon o hindi mangyari sa lahat sa loob ng mahabang panahon. Ipinapalagay na ang pagkalat ng sakit ay: 1 bata sa bawat 20,000 bagong silang. Ngunit ang figure na ito ay napaka-approximate.
Mga sanhi Angelman syndrome
Ang Angelman syndrome ay isang medikal na pangalan para sa isang chromosomal pathology, ngunit ito ay malayo sa isa lamang. Tinatawag ng mga tao ang sakit na ito na doll children syndrome, happy puppet syndrome, Petrushka syndrome, at laughing doll syndrome. Ang mga tao ay gumagawa ng lahat ng uri ng mga pangalan (kung minsan ay nakakasakit sa mga pasyente mismo at sa kanilang mga magulang), ngunit ang isang sakit ay isang sakit, gaano man ito katawa-tawa at anuman ang mga dahilan.
At ang mga dahilan para sa pagbuo ng Angelman syndrome, tulad ng maraming iba pang mga genetic pathologies, sa lahat ng mga kaso ay mga kaguluhan sa istraktura ng isa sa mga chromosome o ang chromosome set bilang isang buo. Ngunit sa aming kaso, ang buong problema ay nasa chromosome 15, na ipinasa mula sa ina. Iyon ay, ang paternal chromosome sa kasong ito ay walang deviations, ngunit ang babae ay sumasailalim sa ilang mga mutasyon.
Ayon sa uri ng chromosomal abnormality, ang Angelman syndrome ay inuri bilang isang chromosomal mutation. Ang ganitong mga mutasyon ay itinuturing na:
- Isang pagtanggal (kawalan ng isang seksyon ng isang chromosome na naglalaman ng isang tiyak na hanay ng mga gene; kung ang isa sa mga gene ay nawawala, pinag-uusapan natin ang tungkol sa isang microdeletion), na resulta ng dalawang break at isang reunion, kapag ang isang seksyon ng orihinal na chromosome ay nawala.
- Pagdoble (ang pagkakaroon ng isang karagdagang seksyon sa isang chromosome na isang kopya ng isang umiiral na), na sa karamihan ng mga kaso ay humahantong sa pagkamatay ng isang tao, at mas madalas sa kawalan ng katabaan.
- Inversion (pagbabaligtad ng isa sa mga seksyon ng chromosome sa pamamagitan ng 180 degrees, ibig sabihin, sa tapat na direksyon, at pagkatapos ay ang mga gene sa loob nito ay matatagpuan sa kabaligtaran na pagkakasunud-sunod), kapag ang mga sirang dulo ng chromosome ay konektado sa isang pagkakasunud-sunod na naiiba mula sa orihinal.
- Pagpasok (kung ang bahagi ng genetic na materyal sa isang chromosome ay wala sa lugar),
- pagsasalin (kung ang isang partikular na seksyon ng isang kromosom ay nakakabit sa isa pang kromosom; ang gayong mutation ay maaaring magkapareho nang walang pagkawala ng mga seksyon).
Ang pagtanggap ng isang mutated chromosome mula sa isang hindi pinaghihinalaang ina, ang bata ay tiyak na ipanganak na may mga abnormalidad. Ang pinakakaraniwang sanhi ng Angelman syndrome ay itinuturing pa rin na isang pagtanggal ng ika-15 chromosome ng ina, kapag ang isang maliit na seksyon ay nawawala. Ang mga hindi gaanong karaniwang mutasyon sa "laughing doll" syndrome ay itinuturing na:
- pagsasalin,
- unipaternal disomy (kung ang bata ay nakatanggap ng isang pares ng chromosome mula sa ama, ang maternal chromosome ay wala),
- mutation ng mga gene sa DNA, na parehong pangunahing gusali (genetic) na materyal at mga tagubilin para sa tamang paggamit nito (sa partikular, mutation ng ube3a gene sa maternal chromosome).
Ang pagkakaroon ng isa sa mga mutasyon na ito sa mga magulang ay isang panganib na kadahilanan para sa pagbuo ng Angelman syndrome sa mga bata. Ngunit hindi lamang ang mga mutasyon ng chromosomal, kundi pati na rin ang mga genomic (na nauugnay sa isang dami ng pagbabago sa mga set ng chromosome at mas karaniwan kaysa sa mga chromosomal) ay maaaring makapukaw ng pag-unlad ng sakit sa isang bata. Kasama sa mga karaniwang genomic mutations ang chromosome trisomy (kung ang chromosome set ng isang tao ay may higit sa 46 chromosome).
Upang lumitaw ang isang patolohiya sa isang bata, hindi kinakailangan para sa mga magulang na magkaroon ng mga abnormalidad ng chromosomal. Gayunpaman, mayroong isang tiyak na porsyento ng mga pasyente na ang sakit ay namamana.
Pathogenesis
Suriin natin nang mas malalim ang biology, o mas tiyak, genetics. Ang genetic na impormasyon ng bawat indibidwal na organismo ng tao ay nakapaloob sa 23 pares ng chromosome. Ang isang chromosome mula sa isang pares ay ipinapasa sa bata mula sa ama, ang isa ay mula sa ina. Ang lahat ng mga pares ng chromosome ay naiiba sa hugis at sukat at nagdadala ng ilang impormasyon. Kaya, ang ika-23 na pares ng chromosome (X at Y chromosome) ay responsable para sa pagbuo ng mga sekswal na katangian ng sanggol (XX - babae, XY - lalaki, habang ang Y chromosome ay maaari lamang matanggap ng bata mula sa ama).
Sa isip, ang isang bata ay tumatanggap ng 46 chromosome mula sa kanyang mga magulang, na bumubuo sa kanyang mga genetic na katangian, na paunang tinutukoy siya bilang isang indibidwal. Ang isang mas malaking bilang ng mga chromosome ay tinatawag na trisomy at itinuturing na isang paglihis mula sa pamantayan. Halimbawa, ang pagkakaroon ng chromosome 47 sa chromosome set (karyotype, pagtukoy ng mga species at indibidwal na katangian) ay nagiging sanhi ng paglitaw ng Down syndrome.
Kung ang mga chromosome ay nabahiran ng isang espesyal na pangulay, pagkatapos ay sa ilalim ng mikroskopyo maaari mong makita ang mga guhitan ng iba't ibang mga kulay sa bawat isa sa kanila. Sa loob ng bawat guhit mayroong isang malaking bilang ng mga gene. Ang lahat ng mga guhit na ito ay binibilang ng mga siyentipiko at may nakapirming lokasyon. Ang kawalan ng isa sa mga guhitan ay itinuturing na isang paglihis mula sa pamantayan. Sa Angelman syndrome, madalas na mapapansin ng isang tao ang kawalan ng mga segment ng maternal chromosome sa pagitan ng q11-q13, na matatagpuan sa mahabang braso, ang bilang ng mga base ng DNA kung saan ay halos 4 milyon lamang.
Ang pangunahing bahagi ng chromosome ay itinuturing na isang hindi kapani-paniwalang mahabang molekula ng DNA na naglalaman ng libu-libong mga gene at sampu at daan-daang milyong nitrogenous base. Kaya, ang chromosome 15, na responsable para sa pagbuo ng Angelman syndrome at ilang iba pa, ay naglalaman ng 1200 genes at humigit-kumulang 100 milyong base. Ang anumang mga kaguluhan sa istraktura ng molekula ng DNA ay tiyak na makakaapekto sa hitsura at pag-unlad ng hinaharap na bata.
Ang genetic na impormasyon na nakapaloob sa mga gene ay na-convert sa protina o RNA. Ang prosesong ito ay tinatawag na gene expression. Sa ganitong paraan, ang genetic na impormasyon na natanggap mula sa mga magulang ay tumatanggap ng parehong anyo at nilalaman, na nakapaloob sa kanilang natatanging babae o lalaki na tagapagmana.
Mayroong isang bilang ng mga pathology na may isang hindi klasikal na uri ng mana, kabilang ang Angelman syndrome, kung saan ang mga gene na natanggap mula sa mga magulang bilang bahagi ng mga ipinares na chromosome ay nagdadala ng isang natatanging imprint ng mga magulang at nagpapakita ng kanilang sarili sa iba't ibang paraan.
Kaya, ang Angelman syndrome ay isang kapansin-pansing halimbawa ng genomic imprinting, kung saan ang expression ng gene sa katawan ng bata ay direktang nakasalalay sa kung saan magulang ang mga alleles ay natanggap mula sa (iba't ibang anyo ng isang gene, na natanggap mula sa ama at ina, na matatagpuan sa magkatulad na mga seksyon ng ipinares na mga chromosome). Iyon ay, ang mga anomalya lamang sa maternal chromosome ay humahantong sa pag-unlad ng sindrom, habang ang mga mutation at structural disorder ng paternal chromosome ay nagdudulot ng ganap na magkakaibang mga pathologies.

Sa patolohiya na ito, may kakulangan ng ilang mga gene sa maternal chromosome o pagkawala/pagbawas sa aktibidad ng mga indibidwal na gene (sa karamihan ng mga kaso, ang ube3a gene, na kasangkot sa metabolismo ng ubiquitin, isang protina na kumokontrol sa pagkasira ng iba pang mga protina). Bilang resulta, ang bata ay nasuri na may mga abnormalidad sa pag-unlad ng kaisipan at mga pisikal na deformidad.
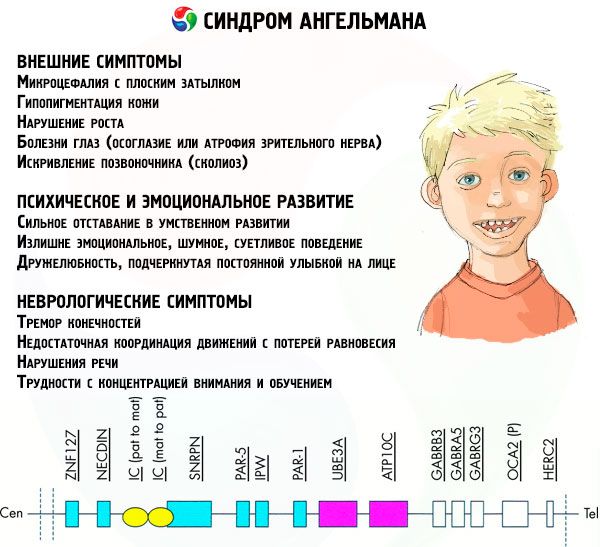
Mga sintomas Angelman syndrome
Ang mga sintomas ng Angelman syndrome ay nakakaapekto sa iba't ibang aspeto ng buhay at pag-unlad ng isang bata: pisikal, neurological, mental. Batay dito, maaaring makilala ang 3 grupo ng mga sintomas na nagpapahiwatig ng pag-unlad ng patolohiya na ito.
- Panlabas o pisikal na mga sintomas:
- isang hindi proporsyonal na maliit na ulo kumpara sa katawan at mga paa, na may normal na laki,
- masyadong malapad ang bibig,
- halos palaging may ngiti sa mukha (na may bukas na bibig),
- kalat-kalat na ngipin,
- makitid na itaas na labi,
- madalas na nakausli ang malawak na dila,
- nakausli sa ibabang panga,
- matulis na baba,
- napakagaan ng balat, madalas na buhok (albinism, na nauugnay sa katotohanan na ang katawan ay hindi gumagawa ng pigment melanin),
- dark spots sa fair skin (hypopigmentation dahil sa hindi sapat na paggawa ng melanin)
- pisikal o panlabas na mga sintomas: mga sakit sa mata tulad ng strabismus o optic nerve atrophy,
- kurbada ng gulugod (scoliosis),
- matigas na binti (kapag naglalakad, ang isang tao ay hindi yumuko sa kanyang mga binti sa tuhod dahil sa mababang kadaliang kumilos ng mga kasukasuan, kaya ang paghahambing sa lakad ng isang manika).
- Mga sintomas na nauugnay sa pag-unlad ng kaisipan at emosyonal:
- malubhang kapansanan sa pag-iisip,
- sobrang emosyonal, maingay, makulit na pag-uugali,
- madalas na pagpalakpak ng mga kamay,
- nagpahayag ng kabaitan, na binibigyang-diin ng patuloy na ngiti sa mukha,
- madalas tumawa ng walang dahilan.
- Mga sintomas ng neurological:
- panginginig ng mga paa,
- hindi sapat na koordinasyon ng mga paggalaw na may pagkawala ng balanse,
- nabawasan ang tono ng kalamnan,
- iba't ibang mga karamdaman sa pagtulog,
- madalas na hysterical fit sa pagkabata,
- mga karamdaman sa pagsasalita (ang bata ay nagsimulang magsalita nang huli, may mahinang kasanayan sa komunikasyon at malabo na pagsasalita),
- hyperactivity laban sa background ng pagtaas ng excitability,
- kahirapan sa pag-concentrate at pag-aaral.
Ngunit ito ay isang pangkalahatang larawan ng sakit. Sa katunayan, ang klinikal na larawan ng Angelman syndrome ay higit sa lahat ay nakasalalay sa yugto ng pag-unlad ng sakit at ang uri ng chromosomal mutation na naging sanhi ng patolohiya. Nangangahulugan ito na ang mga sintomas ng sakit ay maaaring magkakaiba nang malaki sa iba't ibang mga pasyente, na sa loob ng mahabang panahon ay hindi nagpapahintulot sa amin na makilala ang patolohiya mula sa iba na may katulad na klinikal na larawan.
Kabilang sa kabuuang bilang ng mga sintomas, maaari naming i-highlight ang mga katangian ng lahat ng mga pasyente nang walang pagbubukod:
- malubhang kapansanan sa pag-iisip,
- hindi naaangkop na pag-uugali (hindi makatwirang pagtawa, pagtaas ng excitability, mahinang konsentrasyon, estado ng euphoria),
- hindi pag-unlad ng mga kasanayan sa motor,
- mahinang koordinasyon ng mga paggalaw, gait ataxia (hindi pantay na bilis, pag-indayog mula sa gilid sa gilid, atbp.), Panginginig ng mga paa.
- speech development disorder na may nangingibabaw na non-verbal na paraan ng komunikasyon.
Kabilang sa mga sintomas na nakatagpo ng karamihan ng mga pasyente, ang mga sumusunod ay maaaring makilala:
- hindi pagkakatimbang sa pagitan ng ulo at katawan na sanhi ng pagkaantala ng pisikal na pag-unlad,
- sa maraming mga pasyente ang hugis ng bungo ay tulad na ang laki ng utak ay nananatiling mas maliit kaysa sa malusog na tao (microcephaly),
- epileptic seizure bago ang edad na 3 taon na may progresibong pagbaba sa lakas at dalas sa mas matandang edad,
- pagbaluktot ng mga parameter ng EEG (pagbabago at mataas na amplitude ng mga low-frequency na alon).
Ang mga sintomas na ito ay medyo karaniwan, gayunpaman, 20% ng mga pasyente na may Angelman syndrome ay walang mga ito.
Kahit na mas madalas, posible na masuri ang mga pagpapakita ng sakit tulad ng:
- malubhang o banayad na strabismus,
- mahinang kontrol sa paggalaw ng dila, na nagreresulta sa mga pasyente na madalas na lumalabas ang kanilang dila nang walang dahilan,
- kahirapan sa paglunok at pagsuso, lalo na sa maliliit na bata,
- pagkagambala sa pigmentation ng balat at mata,
- nakataas o nakayuko ang mga braso habang naglalakad,
- hyperreflexia,
- mga karamdaman sa pagtulog, lalo na sa pagkabata,
- madalas na paglalaway,
- walang sawang uhaw,
- sobrang aktibong paggalaw ng pagnguya,
- hypersensitivity sa init,
- patag na likod ng ulo,
- nakausli sa ibabang panga,
- makinis na mga palad.
Malaking porsyento ng mga pasyente ang may mga problema sa pag-ihi, na hindi nila kontrolado, may kapansanan sa mahusay na mga kasanayan sa motor, na lumilikha ng mga kahirapan sa pag-aalaga sa sarili at pag-aaral, at labis na timbang. Halos lahat ng mga pasyente ay nakakaranas ng pagdadalaga nang mas huli kaysa sa malusog na mga kapantay.
Ang mga bata na may Angelman syndrome ay nakikita nang mabuti ang pagsasalita sa bibig at naiintindihan ito, ngunit ayaw nilang lumahok sa pag-uusap, nililimitahan ang kanilang pagsasalita sa ilang dosenang mga salita na kinakailangan sa pang-araw-araw na buhay. Gayunpaman, sa pagtanda, ang mga naturang pasyente ay mukhang mas bata kaysa sa kanilang mga kapantay na walang genetic pathologies.
Maraming mga sintomas ng Angelman syndrome ay hindi pare-pareho, kaya ang klinikal na larawan ng sakit ay nagbabago nang malaki sa edad. Ang mga convulsion at epileptic seizure ay nagiging mas madalas o nawawala nang buo, ang pasyente ay nagiging hindi gaanong nasasabik, at ang pagtulog ay bumubuti.
Mga komplikasyon at mga kahihinatnan
Ang Angelman syndrome ay isang malubha, kasalukuyang halos walang lunas na chromosomal pathology na nag-aalis sa mga pasyente ng pagkakataong mamuhay ng normal. Ang magiging buhay ng isang batang may AS ay higit na nakadepende sa uri ng chromosomal abnormality.
Ang pagdoble ng isang chromosome segment ay hindi tugma sa buhay sa karamihan ng mga kaso. At kahit na ang mga naturang pasyente ay hindi namamatay sa pagkabata at umabot sa pagdadalaga, wala silang pagkakataon na magkaanak.
Ang pagtanggal o kawalan ng bahagi ng mga gene na kadalasang nangyayari sa Angelman syndrome ay isang balakid sa pag-aaral ng bata na lumakad at magsalita. Ang ganitong mga bata ay may mas malubhang anyo ng mental retardation, at ang mga epileptic seizure ay nangyayari nang mas madalas, at ang kanilang intensity ay mas malaki kaysa sa mga pasyente na may iba pang mga chromosomal abnormalities.
Kung mayroon lamang isang mutation ng isang gene, na may kaukulang atensyon at diskarte ay maaaring ituro sa bata ang mga pangunahing kaalaman sa pangangalaga sa sarili, komunikasyon at pakikipag-ugnayan sa isang grupo, bagama't siya ay mahuhuli pa rin sa kanyang mga kapantay sa pag-unlad.
Para sa mga batang may Angelman syndrome, na likas na mabait, ang pinakamahalagang bagay ay ang pagmamahal at atensyon ng kanilang mga magulang. Sa kasong ito lamang magbubunga ang edukasyon ng bata, kahit maliit. Siyempre, ang mga pasyente na may AS ay hindi makakapag-aral sa isang regular na paaralan. Kailangan nila ng mga espesyal na klase kung saan tuturuan muna ang mga bata na mag-concentrate, at pagkatapos ay unti-unting bibigyan ng mga pangunahing kaalaman sa paaralan.
Diagnostics Angelman syndrome
Ang Angelman syndrome ay isang congenital developmental pathology. Ngunit dahil sa ilang mga pangyayari, madalas na imposibleng masuri ito sa pagkabata at maagang pagkabata. Ito ay dahil sa hindi tiyak at mahinang pagpapahayag ng mga sintomas sa mga sanggol at batang wala pang 3 taong gulang. At ang paglaganap ng sakit sa ating bansa ay hindi napakalaki na natutunan ng mga doktor na kilalanin ito sa mga kapantay nito.
Ang Angelman syndrome sa mga sanggol ay maaaring magpakita ng sarili bilang nabawasan ang tono ng kalamnan, na nagpapakita ng sarili sa mga problema sa pagpapakain (kahinaan ng pagsuso at paglunok ng reflex), at sa paglaon ay mga paghihirap sa pag-aaral na lumakad (ang gayong mga bata ay nagsimulang maglakad nang maglaon). Ang mga sintomas na ito ay ang mga unang palatandaan ng abnormalidad sa pag-unlad ng sanggol, na maaaring maiugnay sa abnormalidad ng chromosomal. Tanging ang genetic analysis lamang ang makapagpapatunay sa pagpapalagay na ito.
Ang espesyal na atensyon ay binabayaran sa mga bata na ang mga magulang ay may iba't ibang genomic o chromosomal disorder. Pagkatapos ng lahat, ang sakit ay maaaring hindi magpakita mismo sa una, at kung ang patolohiya ay napansin sa oras, sa pamamagitan ng pagsisimulang magtrabaho nang masinsinan sa bata, posible na makamit ang mas malaking tagumpay sa pag-aaral, na nagpapabagal sa pag-unlad ng sakit.
Kung ang mga magulang ay may iba't ibang mga abnormalidad ng chromosomal, ang pagsusuri ng genetic ay isinasagawa kahit na bago ipanganak ang sanggol, dahil ang SA ay isa sa mga pathologies na maaaring makita sa yugto ng embryonic.
Ang koleksyon ng materyal para sa genetic na pananaliksik ay maaaring isagawa sa dalawang paraan:
- invasive (na may isang tiyak na porsyento ng panganib, dahil kinakailangan na tumagos sa matris upang kumuha ng sample ng amniotic fluid),
- non-invasive (pagsusuri ng DNA ng sanggol mula sa dugo ng ina).
Pagkatapos ay isinasagawa ang mga sumusunod na pag-aaral:
- fluorescent in situ hybridization (FISH method) – pagbubuklod ng DNA probe na may label na espesyal na dye sa DNA na pinag-aaralan, na sinusundan ng pagsusuri sa ilalim ng mikroskopyo.
- pagsusuri ng mga mutasyon sa ube3a gene at mga naka-print na gene,
- Pagsusuri ng DNA methylation gamit ang mga espesyal na pamamaraan na ginagamit sa genetika.
Ang mga pagsusuri sa genetiko ay nagbibigay ng medyo tumpak na impormasyon sa kaso ng mga abnormalidad ng chromosomal, na nangangahulugang alam ng mga magulang sa hinaharap kung ano ang ihahanda. Gayunpaman, may mga pagbubukod. Sa isang tiyak na grupo ng mga pasyente, sa pagkakaroon ng lahat ng mga sintomas na nagpapahiwatig ng patolohiya, ang mga resulta ng pagsubok ay nananatiling normal. Iyon ay, ang patolohiya ay maaari lamang makilala sa pamamagitan ng maingat na pagmamasid sa bata mula sa maagang pagkabata: kung paano siya kumakain, kapag nagsimula siyang maglakad at magsalita, kung yumuko siya sa kanyang mga binti kapag naglalakad, atbp.
Bilang karagdagan sa pamamaraan ng FISH, kabilang sa mga instrumental na diagnostic na pamamaraan para sa Angelman syndrome, maaaring isa-isa ng isa ang tomography (CT o MRI), na tumutulong na matukoy ang kondisyon at laki ng utak, at isang electroencephalogram (EEG), na nagpapakita kung paano gumagana ang mga indibidwal na bahagi ng utak.
Ang mga doktor ay karaniwang gumagawa ng pangwakas na pagsusuri sa edad na 3-7 taon, kapag ang pasyente ay mayroon nang karamihan sa mga sintomas at ang dynamics ng pag-unlad ng sakit ay nakikita.
Anong mga pagsubok ang kailangan?
Iba't ibang diagnosis
Ang Angelman syndrome ay isang genetic na patolohiya na halos walang tiyak na mga pagpapakita. Karamihan sa mga sintomas ay maaaring pantay na magpahiwatig ng parehong AS at iba pang genetic pathologies.
Ang pagkakaiba-iba ng diagnosis ng Angelman syndrome ay isinasagawa sa mga sumusunod na pathologies:
- Pitt-Hopkins syndrome (mga pasyente ay nailalarawan sa pamamagitan ng mental retardation, masayang karakter, nakangiti, mayroon silang medyo malaki at malawak na bibig, microcephaly ay nabanggit). Ang pagkakaiba ay ang mga pag-atake ng hyperventilation at pagpigil ng hininga sa isang estado ng paggising.
- Christianson syndrome (mga pasyente ay mga taong may kapansanan sa pag-iisip na may masayang disposisyon, hindi makapagsalita, na nailalarawan sa pamamagitan ng microcephaly, ataxia, convulsions, hindi sinasadyang paggalaw ng kalamnan).
- Mowat-Wilson syndrome (mga sintomas: mental retardation, epileptic seizure, pointed chin, open mouth, happy expression sa mukha, microcephaly). Pagkakaiba: malaking distansya sa pagitan ng mga mata, mga mata na nakahilig sa loob, bilugan na dulo ng ilong, pabalik-balik na auricle.
- Kabuki syndrome (nailalarawan ng banayad hanggang katamtamang pagkaantala sa pag-iisip, mga problema sa pagsasalita at motor, panghihina ng kalamnan, epileptic seizure, microcephaly, mahabang pagitan sa pagitan ng mga kati, at kapansanan sa koordinasyon). Nailalarawan sa pamamagitan ng arched eyebrows, everted lateral part of the lower eyelid, wide-set eyes, long palpebral fissures na may mahaba at makapal na pilikmata.
- Rett syndrome (pagkita ng kaibhan mula sa AS sa mga kababaihan). Mga sintomas: naantala ang pagbuo ng pagsasalita, mga seizure, microcephaly. Ang pagkakaiba ay walang masayang ekspresyon sa mukha, may mga pag-atake ng apnea at apraxia, na umuusad sa paglipas ng panahon.
- Autosomal recessive mental tardation syndrome 38 (mga sintomas: may markang mental retardation na may pagkaantala sa mga kasanayan sa motor at pagsasalita, kahinaan ng kalamnan, mga problema sa pagpapakain sa pagkabata, impulsivity). Ang natatanging tampok ay ang asul na kulay ng iris.
- MECP 2 gene duplication syndrome (pagkaiba mula sa SA sa mga lalaki). Sintomas: malubhang pagkaantala sa pag-iisip, panghihina ng kalamnan mula pagkabata, mga problema sa pagsasalita o kawalan ng pagsasalita, epilepsy. Mga Pagkakaiba: progresibong myopathy, patuloy na umuulit na mga impeksiyon.
- Kleefstra syndrome (mga sintomas: mga problema sa pagsasalita at pag-iisip, kahinaan ng kalamnan, pagkagambala sa pagtulog, kawalan ng atensyon, bukas na bibig, hyperactivity, seizure, ataxia, mga karamdaman sa balanse). Mga natatanging tampok: patag na mukha, maiksing matangos na ilong, malapad na mga mata, malaki ang nakaangat na ibabang labi, agresibong pagsabog.
- Smith-Magenis syndrome (nailalarawan ng mga seizure, mga problema sa pagtulog, mga karamdaman sa pag-unlad ng intelektwal at motor). Kasama sa mga natatanging tampok ang isang malawak at patag na mukha at isang kilalang noo.
- Koolen-de Vries syndrome (banayad hanggang katamtamang pagkaantala sa pag-iisip, panghihina ng kalamnan, mga seizure, pagkamagiliw). Mga tampok na nakikilala: mahabang mukha na may mataas na noo, nakausli na mga tainga, mga slanted na mata, mataas na joint mobility, congenital heart defects.
- Phelan-McDermid syndrome (mga sintomas: mental retardation, speech disorder o kakulangan sa pagsasalita). Mga Pagkakaiba: malalaking kamay na may nabuong mga kalamnan, kahinaan ng kalamnan mula sa kapanganakan, mahinang pagpapawis.
Ang mga pathologies tulad ng adenyl succinate deficiency, autosomal recessive mental retardation syndrome 1, chromosome 2q23.1 duplication syndrome, FOXG1, STXBP1 o MEF2C gene haploinsufficiency syndromes at ilang iba pa ay maaaring "magyabang" ng mga sintomas na katulad ng Angelman syndrome.
Ang gawain ng doktor ay gumawa ng tumpak na pagsusuri, pag-iiba ng Angelman syndrome mula sa mga pathology na may katulad na mga sintomas, at magreseta ng epektibong paggamot na nauugnay sa nasuri na yugto ng sakit.
Sino ang dapat makipag-ugnay?
Paggamot Angelman syndrome
Ang Angelman syndrome ay isa sa mga pathologies kung saan ang gamot ay naghahanap pa rin ng mabisang paggamot. Ang etiological na paggamot ng sakit ay nasa yugto ng pag-unlad ng iba't ibang mga pamamaraan at paraan, na marami sa mga ito ay hindi pa nasusuri sa mga tao. Nangangahulugan ito na sa ngayon ay kailangang limitahan ng mga doktor ang kanilang sarili sa symptomatic therapy, na nakakatulong na kahit papaano ay maibsan ang hindi nakakainggit na sitwasyon ng mga bata at matatanda na may marionette syndrome, na dumaranas ng epileptic seizure, salivation, hypotension at sleep disorders.
Kaya, posible na bawasan ang dalas at lakas ng epileptic seizure sa tulong ng isang maayos na napiling anticonvulsant na gamot. Ngunit ang buong kahirapan ay ang mga seizure sa mga pasyente na may SA ay naiiba sa mga ordinaryong epileptic seizure dahil ang mga ito ay nailalarawan sa pamamagitan ng ilang uri ng mga seizure, na nangangahulugan na ang kondisyon ay maaaring maibsan sa pamamagitan ng pagbibigay ng ilang mga gamot nang sabay-sabay.
Ang pinakasikat na anticonvulsant na ginagamit upang gamutin ang Angelman syndrome ay: valproic acid, topiramate, lamotrigine, levetiracetam, clonazepam at mga gamot batay sa kanila. Ang hindi gaanong karaniwang ginagamit ay mga gamot batay sa carmazepine, phenytoin, phenobarbital, ethosuximide, dahil ang ilan sa mga ito ay maaaring makapukaw ng isang kabalintunaan na epekto na binubuo sa pagpapalakas at pagtaas ng dalas ng mga epileptic seizure. Nangyayari ito kung ang gamot ay ginagamit bilang bahagi ng monotherapy.
Upang gamutin ang drooling, dalawang paraan ang karaniwang ginagamit: panggamot (mga gamot na pumipigil sa produksyon ng laway) at surgical, na kinabibilangan ng muling pagtatanim ng mga salivary duct. Ngunit sa kaso ng SA, ang mga pamamaraang ito ay itinuturing na hindi epektibo, at ang isyu ay nananatiling bukas. Ang mga magulang at yaong nag-aalaga sa mga naturang pasyente ay kailangang magbayad ng espesyal na pansin sa isyung ito, dahil ang mga pasyente mismo ay karaniwang hindi kinokontrol ang paglalaway, at ang ilan ay hindi kayang pangalagaan ang kanilang sarili.
Ang isa pang problema ay ang maikling tagal ng pagtulog. Kadalasan ang mga bata na may Angelman syndrome ay natutulog nang hindi hihigit sa 5 oras, na may negatibong epekto sa paggana ng buong katawan. Madaling nasasabik, aktibong mga bata na mahilig sa mga laro at komunikasyon (kahit na sinusubukan nilang limitahan ang kanilang sarili sa mga pamamaraang hindi pasalita) ay kapansin-pansing pagod sa araw. Upang magkaroon ng isang mahusay na pahinga, ang katawan ay nangangailangan ng isang malalim, buong pagtulog, ngunit ito ay tiyak ang catch.
Mukhang sapat na ang mga gamot na pampakalma (phenothiazines at hindi tipikal na antipsychotics) na nagpapakalma sa sistema ng nerbiyos upang mapabuti ang pagtulog sa mga nasasabik na pasyente. Ngunit sa kaso ng AS, ang paggamit ng mga naturang gamot ay puno ng paglitaw ng mga negatibong epekto. Samakatuwid, mas gusto pa rin ng mga doktor ang banayad na sleeping pills, tulad ng Melatonin (isang natural na hormonal na gamot batay sa sleep hormone), na ibinibigay sa mga pasyente isang oras bago matulog sa halagang 1 tablet, at Diphenhydramine. Ang dalas ng pangangasiwa at dosis nito ay tinutukoy ng doktor depende sa kondisyon at edad ng pasyente.
Minsan ang mga pasyente na may Angelman syndrome ay may mga problema sa panunaw at dumi. Maaari mong pagbutihin ang iyong dumi gamit ang mga laxatives (mas mabuti ang mga herbal).
O maaari mong lapitan ang problema sa ibang paraan, tulad ng ginawa ng mga Amerikanong doktor, batay sa ilang mga paraan ng paggamot sa autism, dahil maraming mga sintomas na katangian ng AS ay katangian din ng autism (impulsivity, hindi sinasadyang paggalaw, paulit-ulit na pagkilos, kakulangan sa atensyon, mga problema sa komunikasyon, atbp.). Nabanggit na ang pagpapakilala ng hormone secretin, na nag-normalize ng panunaw at dumi, ay may positibong epekto sa atensyon ng mga pasyente, at ang oxytocin ay nakakatulong na mapabuti ang mga kakayahan at memorya ng bata, at tamang pag-uugali.
Totoo, ang mga hormone lamang ay hindi sapat, lalo na pagdating sa mga bata. Sa Angelman syndrome, ang therapy sa pag-uugali, makipagtulungan sa isang psychologist at speech therapist (pagtuturo ng mga non-verbal na pamamaraan ng komunikasyon at sign language) ay ipinahiwatig. Ang edukasyon ng naturang mga bata ay dapat na nakabatay sa isang indibidwal na programa na may partisipasyon ng mga espesyal na sinanay na guro, isang psychologist at mga magulang. Sa kasamaang palad, hindi ito posible sa lahat ng dako, at ang mga pamilya ay naiiwan nang mag-isa sa kanilang problema.
Dahil maraming mga batang pasyente na may AS ang dumaranas ng mababang tono ng kalamnan at mga problema sa kasukasuan, maraming atensyon ang binabayaran sa physiotherapy. Kadalasan, ginagamit ng mga doktor ang paggamit ng mga paraffin application, electrophoresis, at magnetic therapy.
Ang aktibong tonic massage at mga espesyal na ehersisyo ng therapeutic physical training ay makakatulong sa maysakit na bata na tumayo sa kanyang mga paa at maglakad nang may kumpiyansa pagkaraan ng ilang sandali. Ang aquagymnastics ay lalong kapaki-pakinabang sa bagay na ito, na inirerekomenda para sa SA sa malamig na tubig. Pinapataas nito ang tono ng kalamnan at tinuturuan ang bata na kontrolin ang kanyang katawan at i-coordinate ang mga paggalaw.
Paggamot ng anticonvulsant
Ang pinaka-mapanganib na sintomas ng Angelman syndrome ay mga seizure na katulad ng sa epilepsy. Ang sintomas na ito ay sinusunod sa 80% ng mga pasyente, na nangangahulugan na ang lahat ng mga ito ay kailangang inireseta ng epektibong anticonvulsant na paggamot.
Ang paggamot sa mga epileptic seizure ay isinasagawa sa tulong ng mga bitamina at anticonvulsant. Sa Angelman syndrome, na sinamahan ng isang convulsive syndrome, ang mga bitamina ng grupo B, pati na rin ang mga bitamina C, D at E ay magiging kapaki-pakinabang. Ngunit ang pagreseta ng bitamina therapy sa iyong sarili sa kasong ito ay lubhang mapanganib, dahil ang hindi makontrol na paggamit ng mga bitamina ay maaaring mabawasan ang pagiging epektibo ng mga antiepileptic na gamot at makapukaw ng bago, mas malala at matagal na mga seizure.
Ang pagpili ng mga anticonvulsant na gamot at ang reseta ng kanilang epektibong dosis ay dapat ding gawin ng isang espesyalistang doktor. Siya rin ang magpapasya kung ang isang gamot ay sapat o kung ang pasyente ay kailangang uminom ng 2 o higit pang mga gamot sa mahabang panahon.
Para sa karamihan ng mga pasyente, ang mga doktor ay nagrereseta ng mga gamot na valproic acid (Valproic acid, Depakine, Convulex, Valparin, atbp.), na pumipigil sa mga seizure at nagpapabuti sa mood at mental na estado ng mga pasyente.
Ang valproic acid ay magagamit sa anyo ng mga tablet, syrup at mga solusyon sa iniksyon. Ang pinakasikat na gamot ay ang prolonged-release na gamot na "Depakine" sa mga tablet at bilang isang solusyon para sa intravenous administration. Ang dosis ng gamot ay tinutukoy ng doktor nang paisa-isa depende sa timbang, edad at kondisyon ng pasyente.
Ang gamot ay kinuha sa panahon ng pagkain 2 hanggang 3 beses sa isang araw. Ang average na pang-araw-araw na dosis ay 20-30 mg bawat 1 kilo ng timbang ng pasyente, ang maximum ay 50 mg/kg bawat araw.
Contraindications para sa paggamit. Huwag gamitin sa kaso ng dysfunction ng atay at pancreas, hemorrhagic diathesis, hepatitis, porphyria at hypersensitivity sa gamot.
Kasama sa mga side effect ang panginginig ng kamay, digestive at stool disorder, at mga pagbabago sa timbang ng katawan.
Ang "Topiramate" ay isa ring gamot na pinili para sa SA. Ginagawa ito sa anyo ng tableta at ginagamit bilang bahagi ng monotherapy at kasama ng iba pang mga gamot.
Paraan ng pangangasiwa at dosis. Dalhin ang mga tablet nang pasalita anuman ang paggamit ng pagkain. Ang paunang pang-araw-araw na dosis para sa mga matatanda ay 25-50 mg, para sa mga bata - 0.5-1 mg/kg. Bawat linggo, ang dosis ay tumataas ayon sa mga tagubilin ng doktor.
Ang gamot ay hindi dapat kunin sa panahon ng pagbubuntis at paggagatas, pati na rin sa kaso ng hypersensitivity sa mga bahagi nito. Ang gamot ay may maraming iba't ibang epekto.
Mga gamot na maaaring ireseta ng doktor para sa Angelman syndrome: Clomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, atbp.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradisyunal na gamot at homeopathy
Ang tradisyunal na gamot, tulad ng mga homeopathic na paghahanda, ay siyempre medyo ligtas, ngunit ang pagiging epektibo ng naturang paggamot para sa Angelman syndrome ay maaaring ituring na kontrobersyal.
Bagama't makakatulong pa rin ang katutubong paggamot sa ilang bagay. Pinag-uusapan natin ang paghinto ng epileptic seizure. Sa bagay na ito, ang herbal na paggamot ay maaaring maging epektibo.
Ang isang mahusay na epekto ay ibinibigay ng isang koleksyon ng gamot batay sa peony, licorice at duckweed (ang mga bahagi ay kinuha sa pantay na dami). Ang mga damo ay kailangang gilingin sa harina. Pagkatapos ng 2 linggo mula sa simula ng pagkuha nito, maaari mong mapansin ang isang makabuluhang pagbaba sa dalas ng mga seizure.
Ang sabaw ng lavender (1 kutsarita bawat baso ng tubig na kumukulo) ay kapaki-pakinabang din para sa mga cramp. Ang halo ay pinakuluan sa loob ng 5 minuto at inilalagay sa loob ng kalahating oras. Ang gamot ay iniinom sa gabi sa loob ng 14 na araw.
Ang isang may tubig (o alkohol) na pagbubuhos ng motherwort ay itinuturing na epektibo para sa mga epileptic seizure.
Sa mga homeopathic na paghahanda para sa pag-iwas sa mga seizure sa Angelman syndrome, maaari kang gumamit ng mga gamot batay sa chamomile at motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Ngunit dapat itong isaalang-alang na ang isang homeopathic na doktor lamang ang maaaring magreseta ng epektibo at ligtas na mga dosis ng mga gamot sa bawat partikular na kaso.
Pag-iwas
Tulad ng malamang na naunawaan na ng mambabasa, hindi pa napipigilan ng gamot ang mga mutation ng gene at iba pang mga abnormalidad ng chromosomal, gayunpaman, pati na rin upang itama ang sitwasyon. Ito ay maaaring mangyari sa sinuman, dahil ang mga batang may Angelman syndrome ay ipinanganak sa malusog na mga magulang, at ang genetika, na kasalukuyang isa sa hindi gaanong pinag-aralan na sangay ng medisina, ay hindi pa maipaliwanag ito.
Ang tanging bagay na maaaring gawin ay gumawa ng isang responsableng diskarte sa pagpaplano ng pagbubuntis, magparehistro at sumailalim sa mga pagsusuri sa oras. Ngunit muli, ang naturang panukala ay magiging mas pang-edukasyon kaysa sa pag-iwas, tulad ng anumang pagsusuri. Ngunit maagang malalaman ng mga kabataang magulang kung ano ang ihahanda, at kung sakaling magkaroon ng positibong sagot, sila ang magpapasiya kung kaya nilang gampanan ang responsibilidad gaya ng pagpapalaki sa isang maysakit na anak.
Pagtataya
Ang pagbabala para sa Angelman syndrome ay nakasalalay sa likas na katangian ng chromosomal abnormality at ang pagiging maagap ng pagtuklas nito. Ang pinakamahirap na tinamaan ay ang mga bata na ang chromosome 15 ay naglalaman ng "mga puwang" sa mga gene (pagtanggal). Ang posibilidad ng mga naturang pasyente na naglalakad at nagsasalita ay napakababa. Ang ibang mga kaso ay maaaring itama sa pamamagitan ng maingat na diskarte at pagmamahal para sa iyong anak.
Sa kasamaang palad, ang mga naturang pasyente ay hindi magagawang maging ganap na mga miyembro ng lipunan, sa kabila ng katotohanan na sila ay malayo sa hangal, naiintindihan nila ang pagsasalita at ang kahulugan nito. Gayunpaman, magkakaroon sila ng mga problema sa komunikasyon sa natitirang bahagi ng kanilang buhay. Ang mga pasyente ay maaaring turuan ng sign language mula pagkabata, ngunit hindi sila maaaring pilitin na makipag-usap gamit ang mga salita. Ang bokabularyo ng mga pasyenteng "nagsasalita" ay limitado sa pinakamababang salita na ginagamit sa pang-araw-araw na buhay (5-15 salita).
Tulad ng para sa pag-asa sa buhay at pangkalahatang kalusugan ng mga pasyente na may Angelman syndrome, ang mga numero dito ay nagbabago sa mga average na halaga. Sa pagtanda, ang mga pasyente ay kadalasang nahaharap sa mga problema sa kalusugan tulad ng scoliosis at labis na katabaan, na, na may tamang diskarte sa paggamot, ay hindi nagbabanta sa buhay.