Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Keratoderma: sanhi, sintomas, diagnosis, paggamot
Huling nasuri: 07.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
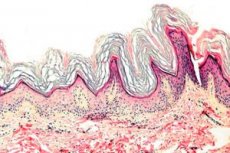
Ang Keratoderma ay isang pangkat ng mga dermatoses na nailalarawan sa pamamagitan ng pagkagambala sa proseso ng keratinization - labis na sungay na pormasyon pangunahin sa mga palad at talampakan.
Ang mga sanhi at pathogenesis ng sakit ay hindi pa ganap na naipaliwanag. Napag-alaman ng pananaliksik na ang mga keratoderma ay sanhi ng mga mutasyon sa mga gene na naka-encode ng keratin 6, 9, 16. Ang kakulangan sa bitamina A, hormonal dysfunctions, pangunahin sa mga glandula ng kasarian, bacterial at viral infection ay may malaking kahalagahan sa pathogenesis. Ang mga ito ay isa sa mga sintomas ng mga namamana na sakit at mga tumor ng mga panloob na organo (parapsoriatic keratodermas).
Mga sintomas. Ginagawa ang pagkakaiba sa pagitan ng diffuse (Unna-Tost keratoderma, Meleda keratoderma, Papillon-Lefevre keratoderma, mutilating keratoderma at mga sindrom na kinabibilangan ng diffuse keratoderma bilang isa sa mga pangunahing sintomas) at focal (disseminated spotted keratoderma ng Fischer-Buschke, acrokeratoelastoidosis ng limitadong keratoderma. Bruhauer-Franzesthesti, linear keratoderma ng Fuchs, atbp.) keratoderma.
Ang Winy-Tost keratoderma (mga kasingkahulugan: congenital ichthyosis ng mga palad at talampakan, Winy-Tost syndrome) ay nakukuha sa autosomal dominant na paraan. Mayroong isang nagkakalat na labis na keratinization ng balat ng mga palad at talampakan (kung minsan lamang ang mga talampakan), na bubuo sa unang dalawang taon ng buhay. Ang proseso ng pathological ng balat ay nagsisimula sa isang bahagyang pampalapot ng balat ng mga palad at talampakan sa anyo ng isang strip ng erythema ng isang mabangis na kulay sa hangganan na may malusog na balat. Sa paglipas ng panahon, lumilitaw ang makinis, madilaw-dilaw na sungay na mga patong sa kanilang ibabaw. Ang sugat ay bihirang kumakalat sa dorsum ng mga pulso o mga daliri. Sa ilang mga pasyente, ang mababaw o malalim na mga bitak ay maaaring mabuo at ang lokal na hyperhidrosis ay nabanggit. Sa pasyenteng naobserbahan ng may-akda, ang tiyuhin sa panig ng ina, kapatid at anak ay nagdusa mula sa Winy-Tost keratoderma.
Ang mga kaso ng pinsala sa mga kuko (pagpapakapal), ngipin, at buhok sa Winy-Tost keratoderma kasama ang iba't ibang mga anomalya ng kalansay at mga pathology ng mga panloob na organo, nervous at endocrine system ay inilarawan.
Histopathology. Ang pagsusuri sa histological ay nagpapakita ng minarkahang hyperkeratosis, granulosis, acanthosis, at maliliit na inflammatory infiltrates sa itaas na dermis. Differential diagnosis. Ang sakit ay dapat na naiiba mula sa iba pang mga uri ng keratoderma.
Ang Meleda keratoderma (mga kasingkahulugan: Meleda disease, congenital progressive acrokeratoma, Siemens' palmoplantar transgradient keratosis, Kogoy's hereditary palmoplantar progressive keratosis) ay minana sa isang autosomal recessive na paraan. Ang form na ito ng keratoderma ay nailalarawan sa pamamagitan ng makapal, dilaw-kayumanggi na mga sungay na layer na may malalim na mga bitak. Ang isang violet-purple border na ilang milimetro ang lapad ay makikita sa mga gilid ng sugat. Ang proseso ay karaniwang kumakalat sa likod ng mga kamay at paa, mga bisig, at mga shins. Karamihan sa mga pasyente ay nakakaranas ng lokal na hyperhidrosis. Kaugnay nito, ang ibabaw ng mga palad at talampakan ay nagiging bahagyang basa-basa at natatakpan ng mga itim na tuldok (mga duct ng glandula ng pawis).
Ang sakit ay maaaring umunlad sa edad na 15-20. Ang mga kuko ay kumakapal at nagiging deformed.
Histopathology. Ang pagsusuri sa histological ay nagpapakita ng hyperkeratosis, kung minsan ay acanthosis, at isang talamak na nagpapasiklab na infiltrate sa papillary dermis.
Differential diagnosis. Ang Melela keratoderma ay dapat na nakikilala mula sa Unna-Tost keratoderma.
Ang Keratoderma Papillon-Lefevre (kasingkahulugan: palmoplantar hyperkeratosis na may periodontitis) ay minana sa isang autosomal recessive na paraan.
Ang sakit ay nagpapakita mismo sa ika-2-3 taon ng buhay. Ang klinikal na larawan ng sakit ay katulad ng sakit ni Melela. Bilang karagdagan, ang mga pagbabago sa mga ngipin ay katangian (mga abnormalidad sa pagsabog ng gatas at permanenteng ngipin na may pag-unlad ng mga karies, gingivitis, mabilis na pag-unlad ng periodontosis na may maagang pagkawala ng ngipin).
Histopathology. Ang pagsusuri sa histological ay nagpapakita ng pampalapot ng lahat ng mga layer ng epidermis, lalo na ang sungay na layer, at hindi gaanong mahalagang mga cellular cluster ng mga lymphocytes at histiocytes sa dermis.
Differential diagnosis. Ang sakit ay dapat na nakikilala mula sa iba pang mga keratodermas. Ang isang mahalagang tampok na nakikilala ay ang katangian ng patolohiya ng ngipin, na hindi matatagpuan sa iba pang mga anyo ng namamana na nagkakalat na keratodermas.
Ang keratoderma mutilans (mga kasingkahulugan: Fonwinkel syndrome, hereditary mutilating keratoma) ay isang uri ng diffuse keratoderma na minana sa isang autosomal dominant na paraan. Ito ay bubuo sa ika-2 taon ng buhay at nailalarawan sa pamamagitan ng nagkakalat na mga malibog na deposito sa balat ng mga palad at talampakan na may hyperhidrosis. Sa paglipas ng panahon, ang mga uka na tulad ng kurdon ay nabubuo sa mga daliri, na humahantong sa mga contracture at kusang pagputol ng mga daliri. Ang follicular keratosis ay ipinahayag sa likod ng mga kamay, pati na rin sa lugar ng siko at mga kasukasuan ng tuhod. Ang mga nail plate ay pinapalitan (kadalasan tulad ng mga salamin sa relo). Ang mga kaso ng hypogonadism, ruby alopecia, pagkawala ng pandinig, pachyonychia ay inilarawan.
Histopathology. Ang pagsusuri sa histological ay nagpapakita ng malubhang hyperkeratosis, granulosis, acanthosis, at maliliit na nagpapasiklab na infiltrates sa dermis, na binubuo ng mga lymphocytes at histiocytes.
Differential diagnosis. Kapag iniiba ang mutilating keratoderma mula sa iba pang anyo ng diffuse keratoderma, ang mutilation effect, na hindi pangkaraniwan para sa iba pang anyo, ay dapat isaalang-alang una sa lahat. Kapag nagsasagawa ng differential diagnostics ng lahat ng anyo ng diffuse keratoderma, kinakailangang tandaan na maaari itong maging isa sa mga pangunahing sintomas ng isang bilang ng mga namamana na sindrom.
Paggamot. Ang Neotigazone ay ipinahiwatig sa pangkalahatang therapy ng keratoderma. Ang dosis ng gamot ay depende sa kalubhaan ng proseso at 0.3-1 mg/kg ng timbang ng pasyente. Sa kawalan ng neotigazone, ang bitamina A ay inirerekomenda sa isang dosis na 100 hanggang 300,000 mg bawat araw sa loob ng mahabang panahon. Ang panlabas na therapy ay binubuo ng paggamit ng mga ointment na may mga aromatic retinoids, keratolytic at steroid agent.
 [
[ Anong bumabagabag sa iyo?
Ano ang kailangang suriin?
Paano masuri?