Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Ang Alkaptonuria ay isang congenital enzyme abnormality
Huling nasuri: 04.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
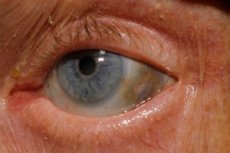
Ang isa sa mga napakabihirang metabolic disorder, ang alkaptonuria, ay tumutukoy sa mga congenital anomalya sa metabolismo ng amino acid tyrosine.
Ang sindrom na ito ay maaari ding tawaging homogentisate oxidase deficiency, homogentisinuria, hereditary ochronosis, o black urine disease.[ 1 ]
Epidemiology
Ayon sa istatistika, hindi hihigit sa siyam na kaso ng alkaptonuria bawat 1 milyong tao. At sa karamihan ng mga bansang Europeo, mayroong isang kaso sa bawat 100-250 thousand live births.
Sa mga bansang Europeo, ang pagbubukod ay ang Slovakia (lalo na ang medyo maliit na hilagang-kanlurang rehiyon), kung saan ang pagkalat ng alkaptonuria ay isang kaso sa bawat 19,000 bagong silang. Ito ay malamang dahil sa ang katunayan na sa mga pamilyang Slovak Roma na naninirahan doon, ang antas ng inbreeding (kasal sa pagitan ng mga magpinsan) ay ang pinakamataas sa Europa: 10-14%. [ 2 ]
Mga sanhi alkaptonuria
Ang eksaktong mga sanhi ng alkaptonuria, bilang isang congenital disorder ng catabolism (metabolic breakdown) ng aromatic (homocyclic) α-amino acid tyrosine, ay naitatag: ang ganitong uri ng metabolic disorder ay bunga ng homozygous o compound heterozygous mutations ng isa sa libu-libong gene sa chromosome 3, mas tiyak sa HG3q3 gene2, mas tiyak, braso ng chromosome. Ang gene na ito ay nag-encode ng mga nucleotide sequence ng liver enzyme homogentisate-1,2-dioxygenase [ 3 ] (tinatawag ding homogentisic acid oxidase o homogentisate oxidase) - isang metalloprotein na naglalaman ng bakal na kinakailangan para sa isa sa mga yugto ng pagkasira ng tyrosine sa katawan. [ 4 ], [ 5 ]
Kaya, ang alkaptonuria ay isang depekto ng enzyme homogentisate-1,2-dioxygenase, o mas tiyak, ang resulta ng genetically determined deficiency o kumpletong kawalan nito. [ 6 ]
Bilang isang congenital enzyme deficiency, ang alkaptonuria ay minana bilang isang autosomal recessive trait, ibig sabihin, para mangyari ang alkaptonuria sa mga bata, ang parehong mga magulang ay dapat magkaroon ng binagong gene para sa enzyme, dahil ang bawat isa sa kanila ay nagpapasa sa bata ng isang kopya lamang ng gene mula sa dalawang magagamit.
Ayon sa pinakabagong data, mayroong higit sa dalawang daang mga variant ng pagbabago ng HGD gene, at ang mga missense mutations, translocation at splicing ay madalas na sinusunod.
Mga kadahilanan ng peligro
Ang tanging panganib na kadahilanan para sa pagbuo ng congenital enzymopathy na ito ay ang pagkakaroon nito sa family history at ang pamana ng dalawang binagong kopya ng HGD gene, kung ang mga magulang ay hindi nagpapakita ng alkaptonuria (ang panganib ng pagpapadala ng anomalya ay 25%), o ang isa sa mga magulang ay may ganitong karamdaman. [ 7 ]
Pathogenesis
Ang tyrosine ay gumaganap ng isang mahalagang papel sa synthesis ng mga protina, ang paggawa ng mga chromoproteins - ang pigment ng balat na melanin, pati na rin ang mga thyroid hormone at catecholamine neurotransmitters.
Ang mekanismo para sa pag-regulate ng dami ng tyrosine sa mga cell ay napaka-kumplikado, at ang katawan ay nag-normalize ng labis na nilalaman nito sa pamamagitan ng pagsira nito. Ang proseso ng tyrosine catabolism, tulad ng lahat ng aromatic amino acids, ay multi-stage at nangyayari sa ilang yugto. Ang bawat yugto ng metabolic breakdown ng tyrosine ay nangyayari sa pakikilahok ng isang tiyak na enzyme at pagbuo ng isang intermediate compound.
Kaya, una ang amino acid ay pinaghiwa-hiwalay sa para-hydroxyphenylpyruvate, na na-convert sa alkaptone - 2,5-dihydroxyphenylacetic o homogentisic acid. Pagkatapos ang alkaptone ay dapat na mabago sa maleacetic acid, ngunit hindi ito nangyayari. [ 8 ]
At ang pathogenesis ng alkaptonuria ay binubuo ng pagtigil ng mga biochemical reaksyon ng tyrosine catabolism sa yugto ng pagbuo ng homogentisic acid: walang kailangan na enzyme upang masira ito - homogentisate oxidase.
Ang homogentisic acid ay hindi ginagamit ng katawan at maaaring maipon sa pamamagitan ng paglabas sa pamamagitan ng mga bato. Bilang karagdagan, ito ay na-oxidized sa benzoquinoacetate (benzoquinoneacetic acid), na, sa pamamagitan ng pagbubuklod sa mga molekula ng mga tisyu at likido ng katawan, ay bumubuo ng mga biopolymer compound na may kulay na tulad ng melanin.
Ang akumulasyon ng mga intermediate na produkto sa tissue ay humahantong sa isang pagkagambala sa collagen structure ng cartilage tissue, na binabawasan ang pagkalastiko nito - na may paglitaw ng maraming mga klinikal na palatandaan ng alkaptonuria at ang pagbuo ng mga komplikasyon.
Mga sintomas alkaptonuria
Ang alkaptonuria sa mga bagong silang at mga sanggol ay nailalarawan sa pamamagitan ng pagdidilim ng ihi. Kapag nalantad sa hangin, ang ihi sa mga lampin, lampin, at damit na panloob ay nagiging dark brown; ito ay dahil sa akumulasyon at pagpapalabas ng homogentisic acid, na na-oxidized sa benzoquinoacetate. [ 9 ]
Sa kawalan ng iba pang mga sintomas, ang alkaptonuria sa mga maliliit na bata ay madalas na hindi nakikilala sa isang napapanahong paraan, dahil ang ihi ay maaaring umitim pagkatapos ng ilang oras ng pag-ihi. Ayon sa ilang data, tanging ang ikalimang bahagi ng mga batang wala pang 12 buwan na ipinanganak na may kakulangan sa enzyme na ito ang natukoy sa mga klinikal na setting. Samakatuwid, napakahalaga para sa mga magulang na bigyang pansin ang pag-aalaga sa kanilang mga sanggol.
Bukod pa rito, ang mga maagang palatandaan ay kinabibilangan ng pigmentation (bluish-gray na kulay) ng sclera ng mga mata at ang mga cartilage ng tainga at ilong, na kadalasang tinatawag na ochronosis.[ 10 ]
Sa paglipas ng panahon, lumilitaw ang iba pang mga sintomas:
- matinding pigmentation ng balat sa cheekbones, armpits at maselang bahagi ng katawan;
- paglamlam ng damit kapag nakikipag-ugnay sa mga pawis na bahagi ng katawan;
- pag-atake ng pangkalahatang kahinaan;
- paos na boses.
Dapat itong isipin na ang alkaptonuria at ochronosis, tulad ng nabanggit sa itaas, ay magkasingkahulugan na mga pangalan para sa parehong disorder ng tyrosine catabolism.
Sakit sa ihi ng maple syrup at alkaptonuria. Ang congenital maple syrup urine disease o leucinosis ay isa ring metabolic disorder, ay may parehong pattern ng mana, at kahit na ang mga mutasyon ay nangyayari sa parehong chromosome, ngunit nakakaapekto sa gene encoding ang enzyme complex ng branched-chain α-keto acid dehydrogenase. Dahil dito, hindi masisira ng katawan ang ilang bahagi ng mga protina, lalo na, ang mga amino acid na leucine, isoleucine at valine. Sa sakit na ito, ang ihi (at earwax) ay may matamis na amoy; bilang karagdagan, ang klinikal na larawan ng ganitong uri ng organic acidemia ay kinabibilangan ng hypopigmentation, pagbabagu-bago sa presyon ng dugo, mga seizure, pagsusuka at pagtatae, pagbaba ng antas ng glucose sa dugo, ketoacidosis, guni-guni, atbp. Ang dami ng namamatay sa mga bata ay medyo mataas; sa mga may sapat na gulang, nang walang paggamot, maaaring mangyari ang koma at kamatayan dahil sa cerebral edema.
Ang albinism at alkaptonuria ay "pinagkakaisa" lamang ng tyrosine. Albinism, kabilang ang oculocutaneous, ay sanhi ng genetic mutations na nakakaapekto sa produksyon ng pigment melanin. Ang mga congenital na pagbabago ay nabanggit sa TYR gene sa chromosome 11 (11q14.3), na nagko-code para sa tyrosinase, isang melanosome enzyme na naglalaman ng tanso na kinakailangan para sa pagbuo ng pigment ng balat batay sa mga produkto ng tyrosine metabolism. Ang sakit na ito ay mas karaniwan kaysa sa alkaptonuria.
Mga komplikasyon at mga kahihinatnan
Dahil sa pagkilos ng mga intermediate metabolites ng tyrosine - homogentisic at benzoquinoneacetic acid - ang mga kahihinatnan at komplikasyon ng alkaptonuria ay lilitaw dahil sa pag-aalis ng mga reaktibong pigmented polymers, ang pagkasira ng collagen fibrils at ang pagkasira ng kondisyon ng cartilage (na may pagbawas sa kanilang pagtutol sa mekanikal na stress).
Sa paglipas ng mga taon, sa pagtanda, ang degenerative arthritis at osteoarthritis ng malalaking joints (hip, sacroiliac at tuhod) ay bubuo; makitid ang mga intervertebral space (lalo na sa lumbar at thoracic spine) - na may calcification at pagbuo ng osteophytes; ang density ng tissue ng subchondral bone plates ay bumababa, at ang pinagbabatayan na mga buto ay maaaring sumailalim sa pathological remodeling na may pagbuo ng mga growths at deformation. [ 11 ]
Ang pinsala sa mga balbula ng puso (aortic at mitral) at coronary arteries ay maaaring maobserbahan - na may mga palatandaan ng coronary heart disease, pati na rin ang pagbuo ng mga bato sa mga bato at prostate gland - dahil sa parehong calcification. [ 12 ], [ 13 ]
Diagnostics alkaptonuria
Karaniwan, ang diagnosis ng congenital metabolic disorder ay batay sa pag-aaral ng mga biological fluid ng katawan.
Batay sa anong mga pagsusuri at reaksyon ang maaaring masuri ang alkaptonuria? Ang mga pagsusuri sa ihi ay kinakailangan upang makita ang homogentisic acid at matukoy ang antas nito (normal - 20-30 mg bawat araw, nakataas - 3-8 g). Ang sample ng ihi ay sinusuri ng gas chromatography o mass spectrometry, gamit ang liquid chromatography; posible ang isang screening test para sa pagkakaroon ng iron chloride sa ihi. [ 14 ]
Mayroon ding paraan para sa mabilis na mga diagnostic - pagtukoy ng alkapton sa mga pinatuyong mantsa ng ihi sa papel (sa pamamagitan ng intensity ng kulay).
Kapag nilinaw ang diagnosis, ang instrumental diagnostics (radiography) ay nagsasangkot ng pagtukoy ng mga radiological sign ng osteoarthritis at iba pang joint pathologies sa mga pasyente.
Ang diagnosis ay nakumpirma ng mga molecular genetic na pamamaraan ng pag-diagnose ng mga namamana na sakit, tulad ng genetic testing at DNA sequencing. [ 15 ]
Iba't ibang diagnosis
Kasama sa differential diagnosis ang hemochromatosis at acute liver failure ng bagong panganak, melaninuria, acute intermittent porphyria, hemophagocytic lymphohistiocytosis, pangunahing mitochondrial pathology, rheumatoid arthritis, ankylosing spondylitis.
Sino ang dapat makipag-ugnay?
Paggamot alkaptonuria
Ang pangunahing paggamot para sa alkaptonuria ay ang oral administration ng malalaking dosis (hindi bababa sa 1000 mg bawat araw) ng ascorbic acid. Sa mga bata, pinapataas nito ang paglabas ng homogentisic acid sa ihi, at sa mga matatanda, binabawasan nito ang nilalaman ng ihi ng derivative nito, benzoquinoneacetic acid, at pinapabagal ang pagbubuklod nito sa mga istruktura ng connective tissue ng mga joints at collagen. [ 16 ]
Sinusuri ng mga klinika sa Kanlurang Europa ang gamot na Nitisinone (Orfalin), isang gamot mula sa pangkat ng mga metabolite na pumipigil sa ikalawang yugto ng tyrosine catabolism: ang pagbabago ng para-hydroxyphenylpyruvate sa homogentisic acid. Gayunpaman, ang paggamit ng pharmacological agent na ito ay humahantong sa akumulasyon ng tyrosine at maaaring magdulot ng matinding side effect, kabilang ang corneal opacity at photophobia, nosebleed at pagdurugo ng tiyan, liver failure, mga pagbabago sa dugo, atbp. Gayunpaman, sa United States, ang Nitisinone ay inaprubahan ng FDA para sa paggamot ng type I tyrosinemia. [17 ], [ 18 ]
Dahil dito, ang paggamot sa physiotherapy - therapy sa ehersisyo upang mapataas ang lakas ng kalamnan at mapabuti ang mobility ng joint, balneotherapy at peloid therapy upang limitahan ang sakit - ay isinasagawa para sa magkasanib na mga problema na dulot ng alkaptonuria.
Bagaman ang tyrosine ay hindi lamang ibinibigay sa pamamagitan ng pagkain ngunit ginawa rin sa katawan, ang mga pasyente na may alkaptonuria ay inirerekomenda na sundin ang isang diyeta na mababa ang protina at limitahan ang pagkonsumo ng mga pagkaing mayaman sa tyrosine, pangunahin ang karne ng baka at baboy, mga produkto ng pagawaan ng gatas (lalo na ang mga keso), legumes, mani at buto.
Pag-iwas
Ang pag-iwas sa mga mutation ng gene ay imposible, ngunit upang maiwasan ang pagsilang ng mga bata na may mataas na panganib ng congenital disorder, mayroong medikal na genetic counseling, na kinakailangan bago ang isang nakaplanong pagbubuntis para sa mga mag-asawa na ang family history ay kinabibilangan ng mga namamana na sakit. [ 19 ]
Pagtataya
Ang mga nakamamatay na kinalabasan mula sa alkaptonuria ay napakabihirang, at ang kamatayan ay maaaring sanhi ng malubhang komplikasyon na kinasasangkutan ng puso at bato. Kaya't ang pangkalahatang pag-asa sa buhay ng mga taong may alkaptonuria ay mabuti.
Ngunit ang kalidad ng buhay ay nabawasan dahil sa matinding sakit sa mga kasukasuan o gulugod na may makabuluhang limitasyon ng kadaliang kumilos, kadalasang umuunlad.