Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Amyloidosis ng atay
Huling nasuri: 07.06.2024

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Ang Amyloidosis ay karaniwang isang systemic, pangkalahatang patolohiya na nailalarawan sa pamamagitan ng akumulasyon ng amyloid (isang tiyak na glycoprotein) sa mga tisyu at kasunod na pagkagambala ng normal na pag-andar ng organ. Ang amyloidosis sa atay ay hindi gaanong karaniwan kaysa sa bato at pali [1] ngunit halos palaging kasama ang sistematikong pinsala sa katawan. Wala sa umiiral na mga diskarte sa imaging maaaring partikular na magpakita ng pagkakaroon ng amyloid. Kahit na pinaghihinalaang klinikal at radiologically, ang diagnosis ng amyloidosis ay nakasalalay sa biopsy ng tisyu upang kumpirmahin ang pagkakaroon ng mga deposito ng amyloid. [3] Ang paggamot ay kumplikado, komprehensibo, at may kasamang immunosuppressive at sintomas na mga hakbang. Sa mga malubhang kaso, maaaring kailanganin ang paglipat ng atay.
Epidemiology
Ang tagumpay ng paggamot ay direktang nakasalalay sa napapanahong pagsusuri ng sakit, na nagiging sanhi ng pagbuo ng isang protina-polysaccharide complex (amyloid) sa iba't ibang mga organo at atay. Tulad ng ipinapakita sa kasanayan, ang amyloidosis ay mahirap ipalagay o pinaghihinalaan, bagaman posible na makilala at kumpirmahin ito. Ang katotohanan ay na sa higit sa 80% ng mga hindi nakikilalang mga kaso, ang sakit ay klinikal na naka-mask sa pamamagitan ng hepatic pathology. Ang pinaka-epektibong pamamaraan ng diagnostic ay biopsy.
Ang atay amyloidosis ay isang rarer na problema kung ihahambing sa renal amyloidosis. Kasabay nito, ang lahat ng mga kaso ng hepatic lesyon ay sinamahan ng mga sugat ng iba pang mga organo. Kadalasan, ang patolohiya ay nakakaapekto sa nakararami na mga istrukturang bahagi ng hepatic triad, na tumutukoy sa minimum at walang katuturan ng sintomas. Ang klinikal at morphological na larawan ng kakulangan sa hepatocellular at hypertension ng portal ay ipinahayag sa nagkakalat at intralobular na uri ng patolohiya.
Ang isang biopsy ng atay ay nabigyang-katwiran kapag ang hepatomegaly ay naroroon nang walang mga nakaraang sintomas ng hepatic at sa kawalan ng nephrotic syndrome.
Ang nagkakalat na pagkakasangkot sa atay ay nakikita sa halos 25% ng mga kaso, at sa 75% ng mga pasyente lamang ang mga portal tract ang apektado.
Ang pangunahing amyloidosis ay nakakaapekto sa atay sa 90% ng mga kaso, habang ang pangalawang amyloidosis ay nakakaapekto sa atay sa 47% lamang ng mga kaso.
Ang nakahiwalay na pagkakasangkot sa atay ay napakabihirang. Ang mga bato (tungkol sa 93%ng mga kaso), pali (72%), puso (57%), pancreas (36%), mga adrenal glandula (29%), bituka at baga (21%bawat isa) ay karaniwang apektado nang magkakasabay.
Ang mga kababaihan ay nakakakuha ng sakit na halos dalawang beses nang madalas bilang mga kalalakihan. Ang average na pag-asa sa buhay ng mga pasyente ng amyloidosis ay 52-64 taon.
Mga sanhi hepatic amyloidosis
Ang Amyloidosis ay nagpapatuloy sa pagbuo at akumulasyon ng isang kumplikadong polysaccharide-protein complex - amyloid - sa tisyu ng atay. Ang problema ng paglitaw ng pangunahing sugat hanggang ngayon ay hindi sapat na pinag-aralan. Tulad ng para sa pangalawang patolohiya, ang hitsura nito ay karaniwang nauugnay sa naturang mga sakit:
- Talamak na nakakahawang proseso (tuberculosis, syphilis, actinomycosis);
- Purulent na nagpapaalab na proseso (microbial endocarditis, osteomyelitis, sakit na bronchiectatic, atbp.);
- Malignant disease (leukemia, visceral cancer, lymphogranulomatosis).
Ang reaktibo na anyo ng amyloidosis ay matatagpuan sa mga pasyente na may magkakasamang atherosclerosis, mga sakit na rheumatologic (sakit ng Bechterew, rheumatoid arthritis), psoriasis, talamak na nagpapaalab at proseso ng multisystem (kabilang ang sarcoidosis). Ang pangunahing mga kadahilanan ng peligro: Hereditary predisposition, cellular immunity disorder, hyperglobulinemia.
Pathogenesis
Mayroong isang bilang ng mga pagpapalagay tungkol sa pinagmulan ng atay amyloidosis. Karamihan sa mga espesyalista ay sumunod sa bersyon ng dysproteinosis, immunologic at mutational na likas na katangian ng sakit, pati na rin ang lokal na cellular genesis. Ang bersyon ng cellular genesis ay nagsasama ng mga pagbabago sa mga reaksyon na nagtatrabaho sa antas ng cellular (pagbuo ng fibrillar precursors ng amyloid sa pamamagitan ng isang kumplikadong macrophage), bagaman ang amyloid ay nabuo at nag-iipon sa labas ng mga istruktura ng cellular.
Ang bersyon ng dysproteinosis ay batay sa katotohanan na ang amyloid ay isang produkto ng hindi tamang metabolismo ng protina. Ang pangunahing link ng pathogenetic ng problema ay namamalagi sa dysproteinemia at hyperfibrinogenemia, na humantong sa akumulasyon ng magaspang na nagkalat na protina at mga fraction ng paraprotein sa plasma.
Ayon sa immunologic na bersyon, ang pagbuo ng amyloid ay sanhi ng reaksyon ng antigen-antibody, kung saan ang mga produktong pagkabulok ng tisyu o mga dayuhang protina ay kumikilos bilang mga antigens. Ang akumulasyon ng amyloid ay matatagpuan higit sa lahat sa lugar ng pagbuo ng antibody at labis na pagkakaroon ng mga antigens.
Ang pinaka-posible na bersyon ng mga siyentipiko ay isinasaalang-alang ang teorya ng mutation, na isinasaalang-alang ang iba't ibang mga kadahilanan ng mutagenic na maaaring humantong sa mga abnormalidad sa synthesis ng protina.
Ang Amyloid ay isang kumplikadong hypoprotein na binubuo ng mga globular at fibrillar protein na sinamahan ng polysaccharides. Ang mga akumulasyon ng amyloid ay nakakaapekto sa intima at adventitia ng vascular network, ang stroma ng mga parenchymatous organo, ang istraktura ng mga glandula, atbp. Ang mga maliliit na akumulasyon ay hindi nagiging sanhi ng mga karamdaman sa pag-andar, ngunit sa matinding pagkakaroon ng amyloid ng pagtaas ng atay sa dami, binabago ang hitsura ng organ, bubuo ng isang kakulangan ng pag-andar.
Ang atay amyloidosis ay nailalarawan sa pamamagitan ng pag-aalis ng mga amyloid fibrils sa puwang ng dysse, na karaniwang nagsisimula sa periportal na rehiyon, bagaman kung minsan ay sentrilobular at maaari ring magdeposito sa hepatic vasculature. [4], [5] Sa mga malubhang kaso, ang pag-aalis ng amyloid ay humahantong sa pagkasayang ng presyon ng mga hepatocytes, na pumipigil sa pagpasa ng apdo, na nagreresulta sa cholestasis, o maaaring hadlangan ang mga sinusoid, na nagreresulta sa portal hypertension. [6], [7], [8]
Mga sintomas hepatic amyloidosis
Ang klinikal na larawan sa atay amyloidosis ay magkakaiba, nakasalalay sa intensity ng amyloid na akumulasyon, sa mga tampok na biochemical nito, tagal ng proseso ng pathologic, ang antas ng pagkasira ng organ at paglabag sa kanilang pagganap na estado.
Sa likas na yugto ng amyloidosis, kapag ang mga akumulasyon ng amyloid sa atay ay maaaring makita lamang sa pamamagitan ng pagsusuri ng mikroskopiko, ang mga unang palatandaan ng sakit ay wala. Sa karagdagang pag-unlad at pagtaas ng functional deficit ng organ, ang symptomatology ay umuusbong.
Unti-unting lumapot ang atay, pinalaki. Ang pamamaraan ng palpation ay maaaring mabago, ngunit makinis at walang sakit na mga hangganan ng organ. Bihirang, ang patolohiya ay sinamahan ng sakit sa lugar ng subcostal sa kanang bahagi, dyspepsia, pagpapalaki ng pali, pag-yellowing ng balat, mauhog lamad at sclerae, hemorrhagic syndrome.
Ang pinaka-katangian na mga sintomas sa atay amyloidosis: [9], [10]
- Ang pag-iipon ng amyloid sa atay ay nagdudulot ng hepatomegaly sa 33-92% ng mga pasyente;
- Banayad na jaundice
- Portal hypertension;
- Katamtaman hanggang sa malubhang cholestasis.
Dahil ang amyloidosis ay bihirang nakakaapekto lamang sa isang organ, ang mga karagdagang symptomatology ay karaniwang naroroon:
- Kapag ang pinsala sa bato ay bubuo ng nephrotic syndrome at arterial hypertension na may karagdagang kabiguan sa bato, edema, kung minsan ang renal vein thrombosis, leukocyturia, hematuria, hypoproteinemia, azotemia at iba pa;
- Kapag apektado ang puso, ang isang kondisyon na katulad ng paghihigpit na cardiomyopathy ay bubuo (mga kaguluhan sa ritmo, cardiomegaly, pagtaas ng kakulangan sa puso, kahinaan at dyspnea, edema, mas madalas - likido na akumulasyon sa tiyan at pleural na lukab, pericarditis);
- Kung ang digestive tract ay apektado, macroglossia, kahinaan at esophageal peristalsis, pagduduwal at heartburn, tibi o pagtatae, atbp ay maaaring mangyari;
- Kapag apektado ang pancreas, ang mga sintomas ng talamak na pancreatitis ay naroroon;
- Kung ang mekanismo ng musculoskeletal ay kasangkot, simetriko polyarthritis, carpal tunnel syndrome, nabuo ang myopathies, at kung ang sistema ng nerbiyos ay apektado, polyneuropathies, paralysis, orthostatic mababang presyon ng dugo, nadagdagan ang pagpapawis, ang demensya ay natagpuan.
Kung ang reaksyon ng pathologic ay kumakalat sa balat, maraming mga waxy plake ang lumilitaw sa mukha, leeg, mga fold ng balat. Ang larawan ng neurodermatitis, pulang squamous fever, scleroderma ay posible.
Ang kumbinasyon ng maraming mga amyloid lesyon at ang iba't ibang mga sintomas na ginagawang pagkilala sa hepatic amyloidosis na mas mahirap at nangangailangan ng isang komprehensibo at kumpletong pagsusuri.
Mga Form
Ayon sa pag-uuri ng WHO, limang uri ng amyloidosis ang nakikilala:
- Al (pangunahing);
- Aa (pangalawang);
- Attr (namamana at senile systemic);
- Aβ2M (sa mga pasyente sa hemodialysis);
- AIAPP (sa mga pasyente na may insulin-independiyenteng diabetes mellitus);
- AB (para sa sakit na Alzheimer);
- AANF (senile atrial amyloidosis).
Mayroong isang lokal na amyloidosis ng atay, ngunit mas madalas na ito ay isang sistematikong sugat, kung saan ang proseso ng pathological ay nagsasangkot din sa mga bato, puso, pali, sistema ng nerbiyos, pati na rin ang iba pang mga organo at tisyu.
Mga komplikasyon at mga kahihinatnan
Ang sistematikong amyloidosis ay unti-unting humahantong sa pagbuo ng mga talamak na proseso ng pathologic na maaaring, sa turn, ay humantong sa kamatayan. Kabilang sa mga pinaka-karaniwang at nagbabanta na mga komplikasyon ay ang mga sumusunod:
- Madalas na nakakahawang (bakterya, viral) na mga pathologies, kabilang ang pneumonias, pyelonephritis, glomerulonephritis;
- Talamak na hepatic at renal failure;
- Talamak na pagkabigo sa puso (maaaring unahan ang myocardial infarction);
- Hemorrhagic stroke.
Ang venous trombosis ay nangyayari bilang isang resulta ng akumulasyon at pag-aalis ng mga protina sa mga venous wall. Ang lumen ng mga apektadong sasakyang-dagat ay nakitid, ang pagkabigo ng organ ay bubuo. Sa paglipas ng panahon, laban sa background ng pangmatagalang hyperproteinemia, ang daluyan ay maaaring ganap na isara. Ang alinman sa mga komplikasyon ay maaaring humantong sa isang hindi kanais-nais na kinalabasan - kamatayan.
Diagnostics hepatic amyloidosis
Kung pinaghihinalaang ang amyloidosis sa atay, ang mga hakbang sa diagnostic ay isinasagawa pagkatapos ng mandatory consultations, parehong gastroenterologist at therapist, at rheumatologist, cardiologist, dermatologist, neurologist, urologist. Mahalaga na komprehensibong suriin ang data ng anamnesis at mga klinikal na pagpapakita, upang magsagawa ng isang komprehensibong laboratoryo at mga instrumental na diagnostic.
Ang mga pagsubok ay kinakailangang isama ang pagsusuri sa ihi at dugo. Sa hepatic amyloidosis, ang isang kumbinasyon ng leukocyturia na may proteinuria at cylindruria ay madalas na matatagpuan, at hypoproteinemia - na may hyperlipidemia, anemia, hyponatremia at hypocalcemia, nabawasan ang bilang ng platelet. Ang mga paraproteins ay napansin sa ihi at serum electrophoresis.
Ang mga instrumental na diagnostic ay kasama ang:
- Ekg, echo;
- Ultrasound ng tiyan;
- X-ray ng tiyan, esophagus;
- Irrigography, barium x-ray;
- Endoscopy.
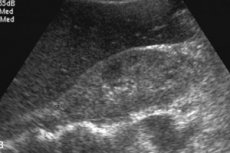
Ang mga natuklasang radiologic ng hepatic amyloidosis ay may kasamang nonspecific hepatomegaly, nadagdagan ang echogenicity sa ultrasound o density sa computed tomography (CT), at nadagdagan ang intensity ng signal ng T1 sa magnetic resonance imaging (MRI). [12] scintigraphy na may mga tagapagpahiwatig na may kaugnayan sa TC-99M ay nagpapakita ng heterogenous na pag-aalsa, ngunit ito ay walang katuturan. [13], [14] Ang gc ay ipinakita upang madagdagan ang higpit ng atay na sinusukat ng elastography; [15], [16], [17] Ngunit may kaunting mga ulat sa kaso. Ang magnetic resonance elastography (MRE) ay kasalukuyang pinaka tumpak na hindi nagsasalakay na pamamaraan upang makita at yugto ng fibrosis ng atay, [18], [19] Ang MRE ay kapaki-pakinabang para sa pagtuklas ng pag-unlad, pagtugon sa paggamot at paghula ng hepatic decompensation sa mga pasyente na may fibrosis ng atay. [20]
Ang Amyloidosis ng atay sa ultrasound ay mahirap matukoy: ang isang pagpapalaki ng organ ay natutukoy, na may pinaka-tiyak na hepatomegaly na higit sa 15 cm. Sa ilalim ng kontrol ng ultrasound, isinasagawa ang isang biopsy, na nagiging isang pagtukoy ng tagapagpahiwatig para sa diagnosis. Gamit ang isang espesyal na karayom, ang isang maliit na halaga ng tisyu ng atay ay kinuha, pagkatapos ito ay marumi na may isang espesyal na pangulay at sinuri sa ilalim ng isang mikroskopyo, na nagbibigay-daan sa iyo upang direktang makita ang mga deposito ng amyloid.
Ang isang tiyak na diagnosis ay ginawa lamang pagkatapos ng pagtuklas ng mga amyloid fibrils sa tisyu ng atay at iba pang mga organo. Ang genetically determined na uri ng amyloidosis ay natutukoy sa pamamagitan ng maingat na genetic-medical na pagsusuri ng pedigree.
Iba't ibang diagnosis
Ang Amyloidosis ay dapat na pinaghihinalaang sa lahat ng mga pasyente na may isang kumbinasyon ng renal proteinuria, paghihigpit na cardiomyopathy, autonomic o peripheral neuropathy, at hepatomyelia, kahit na sa kawalan ng monoclonal paraprotein. Ang pagpapatunay ng uri ng amyloidosis ay napakahalaga dahil ang paggamot ng mga sugat ng iba't ibang mga etiologies ay ibang-iba.
Ang diagnosis ng histological ay nagsasangkot ng paglamlam sa Red Congo na sinusundan ng mikroskopikong pagsusuri sa polarizing light. Maipapayo sa biopsy ng ilang mga sample ng tisyu nang sabay-sabay. Kung ang resulta ng paglamlam ay nagiging positibo, ang immunohistochemical analysis ay isinasagawa gamit ang mga monoclonal antibodies upang ma-precursor ang mga protina upang makilala ang uri ng amyloid.
Ang pagsusuri ng DNA ay isinasagawa upang magkakaiba sa pagitan ng pangunahing amyloidosis at iba't ibang mga pagkakaiba-iba ng genetically determined amyloidosis. Ang mga amyloid fibrils ay maaaring ihiwalay mula sa mga biopsy specimens at sunud-sunod sa mga indibidwal na amino acid.
Karagdagang pag-aaral upang matukoy ang plasma cell dyscrasia:
- Electrophoresis ng serum protein ng dugo at ihi;
- Immunoassay para sa libreng light chain;
- Immunofixation (immunoblotting) ng mga serum protein;
- Bone marrow aspiration at trepanobiopsy.
Ang diagnosis ng amyloidosis ng atay ay isang mahaba at proseso ng masinsinang paggawa, na nangangailangan ng pagtaas ng pansin ng mga espesyalista at kalidad na kagamitan ng mga klinika at laboratoryo.
Sino ang dapat makipag-ugnay?
Paggamot hepatic amyloidosis
Ang mga hakbang sa paggamot ay naglalayong bawasan ang konsentrasyon ng pre-umiiral na mga protina ng amyloid sa dugo (tinanggal ang sanhi ng amyloidosis) at pagsuporta sa sapat na pag-andar ng atay.
Ang pangalawang amyloidosis ay nangangailangan ng pagharang sa nagpapaalab na proseso (sa talamak na nakakahawang at autoimmune pathologies). Sa mga sakit na autoimmune, inirerekomenda ang paggamit ng cytostatics. Upang maalis ang talamak na nakakahawang proseso, ang lugar ng pamamaga ay madalas na tinanggal sa operasyon. Kadalasan ang pamamaraang ito ay maaaring ihinto ang karagdagang pag-unlad ng amyloidosis at pagbutihin ang pag-andar ng atay.
Ang pangunahing amyloidosis ay nangangailangan ng paggamit ng mga gamot na chemopreventive at kung minsan ay ang paglipat ng utak ng buto.
Inirerekomenda ng mga kasalukuyang alituntunin ang pagsasama ng cyclophosphamide, bortezomib, dexamethasone (Cybord), at daratumum bilang first-line therapy sa mga pasyente na bagong nasuri na may AL.
Ang Bortezomib ay isang proteasome inhibitor. Ang mga proteasome ay kasangkot sa pagbabawas ng proteotoxicity at pag-regulate ng mga protina na kumokontrol sa pag-unlad ng cellular at apoptosis. Ang mga cell ng plasma na bumubuo ng amyloid ay partikular na sensitibo sa pagsugpo sa proteasome dahil umaasa sila sa proteasome upang mabawasan ang nakakalason na epekto ng mga light chain at maiwasan ang apoptosis.
Ang Daratumumumab ay isang monoclonal antibody (MAB) na nagbubuklod sa CD38, isang transmembrane glycoprotein na ipinahayag sa ibabaw ng mga cell ng plasma, na nakakaapekto sa apoptosis. Ito ay ang tanging gamot na partikular na naaprubahan para sa paggamot ng AL amyloidosis kapag ginamit sa cybord. Ang pagiging epektibo ng cybord-daratumumumab ay napakataas, na may 78% ng mga pasyente na nakakamit ng isang makabuluhang tugon ng hematologic (tinukoy bilang isang kumpletong tugon o napakahusay na bahagyang tugon). Ang kaligtasan ng median sa maliit na pangkat ng mga pasyente na tumatanggap ng cybord (n = 15) ay 655 araw kumpara sa 178 araw para sa mga pasyente na tumatanggap ng iba pang paggamot na batay sa melphalan-dexamethasone (n = 10). 4
Gayunpaman, ang mga therapy na ito ay may maraming mga epekto, kabilang ang cardiotoxicity, na humahantong sa pangangailangan para sa pagbawas ng dosis o pagsuspinde ng paggamot, at ang paggamit ng iba pang hindi gaanong epektibo ngunit mas matitiis na mga diskarte sa therapeutic.
Ang Isatuximab, isang monoclonal antibody laban sa CD38 na katulad ng daratumumab, ay pinag-aaralan para sa paggamot ng plasma cell dyscrasia na pinagbabatayan ng AL.
Tatlong monoclonal antibodies Birtamimab, CAEL-101 at AT-03 ay kasalukuyang pinag-aaralan para sa pag-alis ng mga amyloid fibrils mula sa mga may sakit na organo. Ang mga resulta ng mga pag-aaral na ito ay maaaring mag-alok ng direktang katibayan para sa hypothesis na sa pamamagitan ng pag-alis ng mga light chain deposition fibrils mula sa mga organo mayroong isang pagpapabuti sa pag-andar ng organ. [21]
Upang suportahan ang pag-andar ng atay, ang mga gamot batay sa urso-deoxycholic acid ay inireseta (halimbawa - ursosan). Ang urso-deoxycholic acid ay tumutulong na patatagin ang mga lamad ng cell, binabawasan ang masamang epekto ng nakakalason na mga fatty acid sa bile stasis na hinimok ng mga deposito ng amyloid, at tumutulong na maibalik ang normal na pag-agos ng apdo.
Bilang karagdagan, ang sintomas ng therapy at suporta para sa paggana ng iba pang mga mahahalagang istruktura tulad ng nervous system, puso, bato, atbp. Ang suporta sa therapy para sa mga pasyente na may hepatic amyloidosis ay may kasamang iba't ibang mga klinikal na aspeto, kabilang ang paggamot ng pagkabigo sa puso, arrhythmias, conduction disorder, thromboembolism at ang magkakasamang pagkakaroon ng aortic stenosis.
Ang iba pang mga paggamot ay nakasalalay sa uri ng amyloidosis at kung aling mga bahagi ng katawan ang apektado. Ang mga paggamot ay maaaring kasama ang: [22]
- Mga gamot na nagpapaginhawa sa mga sintomas, tulad ng mga reliever ng sakit, mga gamot sa pagduduwal, o mga gamot na nagbabawas ng pamamaga (diuretics);
- Mga gamot upang mabawasan ang amyloid;
- Dialysis ng Kidney;
- Transplant ng atay.
Ang atay ay gumagawa ng 95% ng TTR (Transthyretin, isang protina na kasangkot sa transportasyon ng thyroxine (T4) at retinol-nagbubuklod na protina. Ang Transthyretin ay pangunahing na-synthesize sa atay at mayaman sa mga beta strands na may posibilidad na pinagsama-sama sa hindi malulutas na amyloid fibrils) na sinusukat sa suwero. Samakatuwid, ang paglipat ng atay ay may kasaysayan (mula noong 1990) ay iminungkahi bilang first-line therapy upang maalis ang pangunahing mapagkukunan ng amyloidogenic TTR sa mga pasyente na may familial form (ATTRV), samantalang hindi ito ipinahiwatig sa form ng Attr-WT. Ang paglipat ng atay ng mga batang pasyente sa mga unang yugto ng sakit ay nauugnay sa isang mataas na 20-taong rate ng kaligtasan. Ang paglipat ng atay ay lilitaw na mas epektibo sa ilang mga mutasyon at hindi gaanong epektibo sa iba, tulad ng v122i (nauugnay sa cardiomyopathy). Ang pinagsamang paglipat ng atay at puso ay posible rin sa mga batang pasyente na may cardiomyopathy, at ang data ng panitikan sa isang maliit na grupo ng mga pasyente ay nagmumungkahi na ang kumbinasyon na ito ay may mas mahusay na pagbabala kaysa sa paglipat ng puso lamang.
Ang mga pasyente na may hepatic amyloidosis ay kontraindikado na kumukuha ng cardiac glycosides at calcium antagonist tulad ng diltiazem o verapamil, na maaaring makaipon sa amyloid. Ang mga inhibitor ng ACE at beta-adrenoblockers ay ginagamit nang may pag-iingat.
Sa orthostatic hypotension, inireseta ang mineralocorticoids o glucocorticosteroids, na isinasaalang-alang na maaari silang maging sanhi ng pagkabulok ng pagkabigo sa puso. Ang alpha-adrenomimetic midodrine (gutron) ay ginagamit din nang may pag-iingat.
Ang mga anticonvulsant at antidepressant ay angkop sa neuropathies.
Sa ilang mga kaso ng amyloidosis ng atay, dapat isaalang-alang ng mga doktor ang paglipat ng organ.
Pag-iwas
Dahil sa kakulangan ng impormasyon tungkol sa pathogenesis ng atay amyloidosis, ang mga espesyalista ay hindi maaaring bumuo ng isang tiyak na pag-iwas sa sakit. Samakatuwid, ang pangunahing pagsisikap ay nabawasan sa napapanahong pagtuklas at paggamot ng anumang talamak na mga pathologies na maaaring pukawin ang pagbuo ng karamdaman. Kung may mga kaso ng amyloidosis ng anumang lokalisasyon sa pamilya, inirerekomenda na sistematikong bisitahin ang mga doktor para sa mga pagsusuri sa dispensaryo.
Sa pangkalahatan, ang mga hakbang sa pag-iwas ay nabawasan sa napapanahong pag-aalis ng mga nakakahawang sakit, lalo na ang mga may posibilidad na magbago sa isang talamak na proseso. Ito ay tungkol sa pagpigil sa pag-unlad ng tuberculosis, impeksyon sa pulmonary, atbp. Mahalaga sa napapanahong pagtuklas at sapat na paggamot ng mga impeksyon sa streptococcal, na maaaring maging sanhi ng talamak na anyo ng mga proseso ng nagpapaalab na autoimmune. Pinag-uusapan natin ang tungkol sa Scarlatina, streptococcal tonsilitis, atbp.
Kung ang pasyente ay mayroon nang sakit na autoimmune, kung gayon dapat siyang sistematikong kumunsulta sa isang doktor, obserbahan ang aktibidad ng patolohiya, ilapat ang mga kinakailangang gamot tulad ng inireseta ng doktor, ayusin ang mga dosage ayon sa mga indikasyon.
Pagtataya
Ang pagbabala para sa mga pasyente na may hepatic amyloidosis ay hindi kanais-nais. Ang sakit ay tumataas nang dahan-dahan ngunit patuloy na, na sa kalaunan ay nagiging sanhi ng disfunction ng mga apektadong organo at nakamamatay na kinalabasan - lalo na, dahil sa pagkabigo ng organ.
Ang mga pasyente na may sistematikong patolohiya ay pangunahing namatay bilang isang resulta ng pag-unlad ng talamak na pagkabigo sa bato, bagaman sa ilang mga kaso hemodialysis o patuloy na ambulatory peritoneal dialysis ay nagpapabuti sa pagbabala ng mga naturang pasyente. Ang rate ng kaligtasan ng mga pasyente sa hemodialysis, anuman ang uri nito, ay maihahambing sa mga taong may iba pang mga sistematikong pathologies at diabetes mellitus.
Ang pangunahing sanhi ng kamatayan sa panahon ng hemodialysis ay ang pagbuo ng mga komplikasyon mula sa cardiovascular system.
Ang paglipat ng atay ay matagal nang itinuturing na isa sa mga pangunahing pamamaraan ng paggamot ng sakit, at ang pinaka-optimistikong mga rate ng kaligtasan ng buhay ay sinusunod sa mga pasyente na ang edad ay hindi lalampas sa 50 taon (sa kondisyon na ang proseso ng pathological ay maikli ang buhay at ang index ng mass ng katawan ay normal). Ang mga pasyente na may atay amyloidosis na sinamahan ng peripheral neuropathy ay medyo mas masahol na pagbabala.