Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Charcot-Marie-Tooth disease.
Huling nasuri: 12.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Peroneal muscular atrophy, Charcot-Marie-Tooth syndrome o sakit ay isang pangkat ng mga malalang sakit na namamana na may pinsala sa peripheral nerves.
Ayon sa ICD-10, sa seksyon ng mga sakit ng nervous system, ang code para sa sakit na ito ay G60.0 (hereditary motor at sensory neuropathy). Kasama rin ito sa listahan ng mga sakit sa ulila.
Epidemiology
Ayon sa mga klinikal na istatistika, ang pagkalat ng lahat ng uri ng sakit na Charcot-Marie-Tooth sa bawat 100 libong populasyon ay 19 na kaso (ayon sa iba pang mga mapagkukunan, isang kaso bawat 2.5-10 libo ng populasyon).
Ang uri ng CMT 1 ay tumutukoy sa halos dalawang-katlo ng mga kaso (isang kaso bawat 5,000 hanggang 7,000 populasyon), at halos 70% ng mga ito ay nauugnay sa pagdoble ng gene ng PMP22. Mahigit sa 1.2 milyong tao sa buong mundo ang dumaranas ng ganitong uri ng sakit.
Ang saklaw ng CMT type 4 ay tinatantya sa 1-5 kaso bawat 10,000 bata. [ 1 ]
Mga sanhi Charcot-Marie-Tooth disease
Ayon sa pag-uuri ng polyneuropathic syndromes, ang peroneal (fibular) muscular atrophy, Charcot-Marie-Tooth neural amyotrophy o Charcot-Marie-Tooth disease (dinaglat bilang CMT) ay tumutukoy sa genetically determined motor-sensory polyneuropathies. [ 2 ]
Iyon ay, ang mga sanhi ng paglitaw nito ay genetic mutations. At depende sa likas na katangian ng genetic deviations, ang mga pangunahing uri o uri ng sindrom na ito ay nakikilala: demyelinating at axonal. Kasama sa unang grupo ang uri ng sakit na Charcot-Marie-Tooth 1 (CMT1), na nangyayari dahil sa pagdoble ng PMP22 gene sa chromosome 17, na nagko-code para sa transmembrane peripheral myelin protein 22. Bilang resulta, ang segmental demyelination ng axon sheath (mga proseso ng nerve cell) at ang pagbaba sa bilis ng nerve signal conduction. Bilang karagdagan, ang mga mutasyon ay maaaring nasa ilang iba pang mga gene.
Ang axonal form ay Charcot-Marie-Tooth disease type 2 (CMT2), na nakakaapekto sa mga axon mismo at nauugnay sa mga pathological na pagbabago sa MFN2 gene sa locus 1p36.22, na naka-encode ng membrane protein mitofusin-2, na kinakailangan para sa mitochondria fusion at ang pagbuo ng functional mitochondrial networks sa loob ng peripheral nerve cells. Mayroong higit sa isang dosenang mga subtype ng CMT2 (na may mga mutasyon sa mga partikular na gene).
Dapat pansinin na sa kasalukuyan higit sa isang daang mga gene ang natukoy na ang pinsala, na ipinadala sa pamamagitan ng mana, ay nagiging sanhi ng iba't ibang mga subtype ng sakit na Charcot-Marie-Tooth. Halimbawa, ang mga mutasyon sa gene ng RAB7 ay humahantong sa uri ng CMT 2B; Ang pagbabago ng gene ng SH3TC2 (pag-encode ng isa sa mga protina ng lamad ng mga selula ng Schwann) ay nagiging sanhi ng uri ng CMT 4C, na nagpapakita ng sarili sa pagkabata at nailalarawan sa pamamagitan ng demyelination ng motor at sensory neuron (mayroong isang dosenang at kalahating anyo ng uri 4 ng sakit na ito).
Ang isang bihirang uri 3 CMT (tinatawag na Dejerine-Sottas syndrome) ay nagsisimulang bumuo sa maagang pagkabata at sanhi ng mga mutasyon sa PMP22, MPZ, EGR2, at iba pang mga gene.
Kapag ang CMT type 5 ay nangyayari sa edad na 5-12 taon, hindi lamang ang motor neuropathy (sa anyo ng spastic paraparesis ng mas mababang paa't kamay) ay sinusunod, kundi pati na rin ang pinsala sa optic at auditory nerves.
Ang kahinaan ng kalamnan at optic atrophy (na may pagkawala ng paningin) pati na rin ang mga problema sa balanse ay tipikal ng CMT type 6. At sa type 7 Charcot-Marie-Tooth disease ay hindi lamang motor-sensory neuropathy, kundi pati na rin ang isang retinal disease sa anyo ng retinitis pigmentosa.
Mas karaniwan sa mga lalaki, ang X-linked CMT o Charcot-Marie-Tooth disease na may tetraparesis ng mga limbs (kahinaan ng paggalaw ng parehong mga braso at binti) ay isang uri ng demyelinating at pinaniniwalaang nagreresulta mula sa isang mutation sa GJB1 gene sa mahabang braso ng X chromosome, na nagko-encode ng connexin 32 at protina ng mga selulang transregulate ng oligondrondrmembrane, Schodendrondromembrane. ang paghahatid ng mga signal ng nerve. [ 3 ]
Mga kadahilanan ng peligro
Ang pangunahing kadahilanan ng panganib para sa CMT ay isang kasaysayan ng pamilya ng sakit, iyon ay, sa malapit na kamag-anak.
Ayon sa mga geneticist, kung ang parehong mga magulang ay carrier ng autosomal recessive gene para sa sakit na Charcot-Marie-Tooth, ang panganib na magkaroon ng isang bata na magkakaroon ng sakit na ito ay 25%. At ang panganib na ang bata ay magiging carrier ng gene na ito (ngunit hindi magkakaroon ng anumang sintomas) ay tinatantya sa 50%.
Sa kaso ng X-linked inheritance (kapag ang mutated gene ay nasa X chromosome ng babae), may 50% na panganib na ipapasa ng ina ang gene sa kanyang anak, na magkakaroon ng CMT. Ang sakit ay hindi maaaring mangyari kapag ang isang babaeng anak ay ipinanganak, ngunit ang mga anak na lalaki (apo) ng anak na babae ay maaaring magmana ng may sira na gene, at ang sakit ay bubuo.
Pathogenesis
Sa anumang uri ng sakit na Charcot-Marie-Tooth, ang pathogenesis nito ay sanhi ng namamana na anomalya ng peripheral nerves: motor (movement) at sensory (sensitive).
Kung ang uri ng CMT ay demyelinating, kung gayon ang pagkasira o depekto ng myelin sheath na nagpoprotekta sa mga axon ng peripheral nerves ay humahantong sa isang pagbagal sa paghahatid ng mga nerve impulses sa peripheral nervous system - sa pagitan ng utak, kalamnan at pandama na organo.
Sa uri ng axonal ng sakit, ang mga axon mismo ay apektado, na negatibong nakakaapekto sa lakas ng mga signal ng nerve, na hindi sapat para sa buong pagpapasigla ng mga kalamnan at pandama na organo.
Basahin din:
Paano naililipat ang Charcot-Marie-Tooth syndrome? Ang mga may sira na gene ay maaaring mamana sa isang autosomal dominant o autosomal recessive na paraan.
Ang pinakakaraniwang uri, ang autosomal dominant inheritance, ay nangyayari kapag mayroong isang kopya ng mutated gene (na dala ng isa sa mga magulang). At ang posibilidad na maipasa ang CMT sa bawat batang ipinanganak ay tinatayang nasa 50%. [ 4 ]
Sa autosomal recessive inheritance, dalawang kopya ng depektong gene (isa mula sa bawat magulang na hindi nagpapakita ng anumang mga palatandaan ng sakit) ay kailangan para sa pag-unlad ng sakit.
Sa 40-50% ng mga kaso, nangyayari ang autosomal dominant hereditary demyelination, ibig sabihin, CMT type 1; sa 12-26% ng mga kaso, axonal CMT, ie type 2. At sa 10-15% ng mga kaso, ang X-linked inheritance ay sinusunod. [ 5 ]
Mga sintomas Charcot-Marie-Tooth disease
Karaniwan, ang mga unang palatandaan ng sakit na ito ay nagsisimulang lumitaw sa pagkabata at pagbibinata at unti-unting umuunlad sa buong buhay, bagaman ang sindrom ay maaaring makilala ang sarili sa ibang pagkakataon. Ang kumbinasyon ng mga sintomas ay variable, at ang rate ng pag-unlad ng sakit, pati na rin ang kalubhaan nito, ay imposible upang mahulaan.
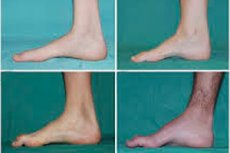
Ang mga tipikal na sintomas ng paunang yugto ay kinabibilangan ng pagtaas ng pangkalahatang pagkapagod; nabawasan ang tono (kahinaan) ng mga kalamnan ng paa, bukung-bukong at shins; kakulangan ng reflexes. Ito ay nagpapahirap sa paggalaw ng paa at humahantong sa dysbasia (gait disturbance) sa anyo ng isang mas mataas na pagtaas ng mga binti, madalas na may madalas na paglalakbay at pagbagsak. Ang mga senyales ng sakit na Charcot-Marie-Tooth sa isang maliit na bata ay maaaring kabilangan ng malinaw na pagka-clumsiness at kahirapan sa paglalakad na hindi karaniwan sa edad na nauugnay sa bilateral foot drop. Ang mga deformidad ng paa ay katangian din: mataas na arko (hollow foot) o matinding flat feet, hubog (hugis-martilyo) na mga daliri.
Sa kaso ng paglalakad sa mga daliri sa paa laban sa background ng hypotonia ng kalamnan, ang isang neurologist ay maaaring maghinala na ang bata ay may uri ng 4 CMT, kung saan ang mga bata ay maaaring hindi makalakad sa pagbibinata.
Habang lumalaki ang sakit, ang pagkasayang ng kalamnan at panghihina ay kumakalat sa itaas na mga paa't kamay, na nagpapahirap sa paggawa ng mga mahusay na kasanayan sa motor at magsagawa ng mga normal na gawain sa kamay. Ang nabawasan na mga pandamdam na sensasyon at ang kakayahang makaramdam ng init at lamig, pati na rin ang pamamanhid sa mga paa at kamay, ay nagpapahiwatig ng pinsala sa mga axon ng sensory nerves.
Sa Charcot-Marie-Tooth disease type 3 at 6 na nagpapakita sa pagkabata, ang sensory ataxia (impaired coordination of movements and balance), muscle twitching at tremor, pinsala sa facial nerve, optic nerve atrophy na may nystagmus, at pandinig ay sinusunod.
Sa mga susunod na yugto, maaaring magkaroon ng hindi makontrol na pagyanig (panginginig) at madalas na pag-cramp ng kalamnan; ang mga problema sa paggalaw ay maaaring humantong sa pag-unlad ng sakit: kalamnan, kasukasuan, neuropathic.
Mga komplikasyon at mga kahihinatnan
Ang sakit na Charcot-Marie-Tooth ay maaaring magkaroon ng mga komplikasyon at kahihinatnan tulad ng:
- mas madalas na sprains at fractures;
- contractures na nauugnay sa pagpapaikli ng periarticular kalamnan at tendons;
- scoliosis (kurbada ng gulugod);
- mga problema sa paghinga - kapag ang mga nerve fibers na nagpapapasok sa mga kalamnan ng diaphragm ay nasira:
- pagkawala ng kakayahang kumilos nang nakapag-iisa.
Diagnostics Charcot-Marie-Tooth disease
Kasama sa diagnosis ang klinikal na pagsusuri, anamnesis (kabilang ang family history), neurological at systemic na pagsusuri.
Isinasagawa ang mga pagsusuri upang suriin ang saklaw ng paggalaw, sensitivity at tendon reflexes. Maaaring masuri ang pagpapadaloy ng nerbiyos gamit ang instrumental diagnostics – electromyography o electroneuromyography. Maaaring kailanganin din ang ultratunog o MRI. [ 6 ]
Ang genetic o DNA testing upang makita ang pinakakaraniwang genetic mutations na nagdudulot ng CMT sa isang sample ng dugo ay limitado dahil ang mga pagsusuri sa DNA ay kasalukuyang hindi magagamit para sa lahat ng uri ng sakit. Para sa higit pang impormasyon, tingnan ang genetic testing
Sa ilang mga kaso, ang isang biopsy ng peripheral nerve (karaniwan ay ang sural nerve) ay isinasagawa.
Iba't ibang diagnosis
Kasama sa differential diagnosis ang iba pang peripheral neuropathies, Duchenne muscular dystrophy, myelopathic at myasthenic syndromes, diabetic neuropathy, myelopathies sa multiple at amyotrophic lateral sclerosis, Guillain-Barré syndrome, trauma sa peroneal nerve at ang pagkasayang nito (kabilang ang kapag naipit sa pagitan ng lumbar disc o thalamus), pati na rin ang mga epekto sa pagitan ng lumbar disc o thalamus. chemotherapy (sa panahon ng paggamot na may mga cytostatics tulad ng Vincristine o Paclitaxel). [ 7 ]
Sino ang dapat makipag-ugnay?
Paggamot Charcot-Marie-Tooth disease
Sa ngayon, ang paggamot para sa namamana na sakit na ito ay kinabibilangan ng exercise therapy (na naglalayong palakasin at palakihin ang mga kalamnan); occupational therapy (na tumutulong sa mga pasyente na may kahinaan sa kalamnan sa mga kamay); at ang paggamit ng mga orthopedic device upang mapadali ang paglalakad. Kung kinakailangan, inireseta ang mga painkiller o anticonvulsant. [ 8 ]
Sa mga kaso ng matinding flat feet, maaaring isagawa ang osteotomy, at sa kaso ng deformation ng takong, ipinahiwatig ang surgical correction - arthrodesis. [ 9 ]
Ang pananaliksik ay isinasagawa sa parehong genetic na bahagi ng sakit at mga paraan ng paggamot nito. Ang paggamit ng mga stem cell, ilang hormone, lecithin o ascorbic acid ay hindi pa nagbubunga ng mga positibong resulta.
Ngunit salamat sa kamakailang pananaliksik, may bagong maaaring lumitaw sa paggamot ng sakit na Charcot-Marie-Tooth sa malapit na hinaharap. Kaya, mula noong 2014, ang Pranses na kumpanya na Pharnext ay umuunlad, at mula noong kalagitnaan ng 2019, ang mga klinikal na pagsubok ay isinasagawa para sa gamot na PXT3003 para sa paggamot ng CMT type 1 sa mga matatanda, na pinipigilan ang pagtaas ng pagpapahayag ng gene ng PMP22, nagpapabuti sa myelination ng peripheral nerves at nagpapagaan ng mga sintomas ng neuromuscular.
Ang mga espesyalista mula sa kumpanyang medikal na Sarepta Therapeutics (USA) ay nagtatrabaho sa paglikha ng gene therapy para sa type 1 na sakit na Charcot-Marie-Tooth. Ang therapy na ito ay gagamit ng hindi nakakapinsalang adeno-associated virus (AAV) ng Dependovirus genus na may linear na single-stranded DNA genome, na maglilipat ng NTF3 gene sa katawan, na nag-e-encode ng neurotrophin-3 (NT-3) na protina na kinakailangan para sa paggana ng Schwann nerve cells.
Si Helixmith ay magsisimula ng mga klinikal na pagsubok ng South Korean-developed gene therapy na Engensis (VM202) sa pagtatapos ng 2020 upang gamutin ang mga sintomas ng kalamnan sa CMT type 1. [ 10 ]
Pag-iwas
Ang pag-iwas sa CMT ay maaaring genetic counseling ng mga magulang sa hinaharap, lalo na kung ang isang tao sa mag-asawa ay may family history ng sakit. Gayunpaman, ang mga kaso ng de novo point gene mutations ay natukoy, iyon ay, sa kawalan ng sakit sa family history.
Sa panahon ng pagbubuntis, ang posibilidad ng sakit na Charcot-Marie-Tooth sa hinaharap na bata ay maaaring masuri ng chorionic villus biopsy (mula 10 hanggang 13 na linggo ng pagbubuntis), pati na rin ang pagsusuri ng amniotic fluid (sa 15-18 na linggo).
Pagtataya
Sa pangkalahatan, ang pagbabala para sa iba't ibang uri ng sakit na Charcot-Marie-Tooth ay nakasalalay sa klinikal na kalubhaan, ngunit sa lahat ng mga kaso ang sakit ay umuunlad nang mabagal. Maraming mga pasyente ang may mga kapansanan, bagaman hindi nito binabawasan ang pag-asa sa buhay.