Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Cystic fibrosis
Huling nasuri: 04.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Ang cystic fibrosis ay isang genetic autosomal recessive monogenic disease na nailalarawan sa pamamagitan ng isang disorder ng pagtatago ng mga exocrine glandula ng mga mahahalagang organo na may pinsala lalo na sa respiratory at digestive system, malubhang kurso at hindi kanais-nais na pagbabala.
 [ 1 ]
[ 1 ]
Epidemiology
Ang saklaw ng cystic fibrosis ay nagbabago sa pagitan ng 1:2,500 at 1:4,600 na bagong silang. Bawat taon, humigit-kumulang 45,000 katao na may cystic fibrosis ang ipinanganak sa buong mundo. Ang saklaw ng cystic fibrosis gene carrier ay 3-4%, na may humigit-kumulang 275 milyong tao sa buong mundo ang mga carrier ng gene na ito, kung saan humigit-kumulang 5 milyon ang nakatira sa Russia at humigit-kumulang 12.5 milyon sa mga bansang CIS.
Mga sanhi cystic fibrosis
Ang cystic fibrosis ay ipinapadala sa isang autosomal recessive na paraan. Ang cystic fibrosis gene ay matatagpuan sa autosome 7, naglalaman ng 27 exon at binubuo ng 250,000 pares ng nucleotide.
Ang isang gene ay maaaring magkaroon ng maraming mutasyon, na ang bawat isa ay tiyak sa isang partikular na populasyon o heyograpikong rehiyon. Mahigit sa 520 mutations ang inilarawan, ang pinakakaraniwan ay delta-P-508, ibig sabihin, isang pagpapalit ng amino acid phenylalanine sa posisyon 508.
Pathogenesis
Ang mga mutasyon sa cystic fibrosis gene ay nakakagambala sa istraktura at paggana ng isang protina na tinatawag na CFTR (cystic fibrosis transmembrane regulator). Ang protina na ito ay gumaganap bilang isang chloride channel na kasangkot sa pagpapalitan ng tubig-electrolyte ng mga epithelial cells ng bronchopulmonary system, gastrointestinal tract, pancreas, atay, at reproductive system. Bilang resulta ng pagkagambala sa pag-andar at istraktura ng protina ng CFTR, ang mga chloride ions Cl - ay naipon sa loob ng cell. Ito ay humahantong sa isang pagbabago sa mga potensyal na elektrikal sa lumen ng excretory ducts, na pinapadali ang daloy ng malalaking halaga ng sodium ions (Na + ) mula sa lumen ng duct papunta sa cell at higit na pinahusay ang pagsipsip ng tubig mula sa pericellular space.
Bilang resulta ng mga pagbabagong ito, ang pagtatago ng karamihan sa mga glandula ng exocrine ay lumalapot, ang paglisan nito ay nagambala, na humahantong sa binibigkas na pangalawang mga karamdaman sa mga organo at sistema, na pinaka-binibigkas sa bronchopulmonary at digestive system.
Ang isang talamak na nagpapaalab na proseso ng iba't ibang intensity ay bubuo sa bronchi, ang pag-andar ng ciliated epithelium ay matalim na nagambala, ang plema ay nagiging napaka-malapot, makapal, napakahirap na lumikas, ang pagwawalang-kilos nito ay sinusunod, ang bronchiolo- at bronchiectasis ay nabuo, na sa paglipas ng panahon ay nagiging mas karaniwan. Ang mga pagbabagong ito ay humahantong sa pagtaas ng hypoxia at pagbuo ng talamak na sakit sa puso sa baga.
Ang mga pasyente na may cystic fibrosis ay lubhang predisposed sa pagbuo ng talamak na pamamaga sa bronchopulmonary system. Ito ay dahil sa binibigkas na mga kaguluhan sa lokal na bronchopulmonary defense system (nabawasan ang mga antas ng IgA, interferon, phagocytic function ng alveolar macrophage at leukocytes).
Ang mga alveolar macrophage ay may malaking papel sa pagbuo ng talamak na pamamaga sa bronchopulmonary system. Gumagawa sila ng malalaking halaga ng IL-8, na lubhang nagpapataas ng neutrophil chemotaxis sa bronchial tree. Naiipon ang mga neutrophil sa malalaking dami sa bronchi at, kasama ng mga epithelial cells, ay naglalabas ng maraming proinflammatory cytokine, kabilang ang IL-1, 8, 6, tumor necrosis factor, at leukotrienes.
Ang isang mahalagang papel sa pathogenesis ng pinsala sa bronchopulmonary system ay nilalaro din ng mataas na aktibidad ng enzyme elastase. Ang isang pagkakaiba ay ginawa sa pagitan ng exogenous at endogenous elastase. Ang una ay ginawa ng bacterial flora (lalo na Pseudomonas aeruginosa), ang pangalawa - ng neutrophilic leukocytes. Sinisira ng Elastase ang epithelium at iba pang mga elemento ng istruktura ng bronchi, na nag-aambag sa karagdagang pagkagambala sa transportasyon ng mucociliary at ang mabilis na pagbuo ng bronchiectasis.
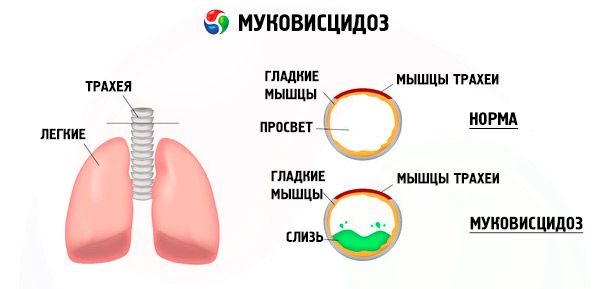
Ang mga neutrophilic leukocytes ay naglalabas din ng iba pang mga proteolytic enzymes. Ang Alpha-1-antipyrsin at secretory inhibitor ng leukoproteases ay humahadlang sa impluwensya ng proteolytic enzymes at, samakatuwid, pinoprotektahan ang bronchopulmonary system mula sa kanilang nakakapinsalang impluwensya. Gayunpaman, sa kasamaang-palad, sa mga pasyente na may cystic fibrosis, ang mga proteksiyong salik na ito ay pinipigilan ng isang malaking halaga ng neutrophilic protease.
Ang lahat ng mga pangyayaring ito ay nag-aambag sa pagpapakilala ng impeksiyon sa bronchopulmonary system at pag-unlad ng talamak na purulent bronchitis. Bilang karagdagan, dapat itong isaalang-alang na ang may sira na protina na naka-encode ng cystic fibrosis gene ay nagbabago sa functional na estado ng bronchial epithelium, na pinapaboran ang pagdirikit ng bakterya sa bronchial epithelium, lalo na ang Pseudomonas aeruginosa.
Kasama ng patolohiya ng bronchopulmonary system, ang cystic fibrosis ay nagdudulot din ng matinding pinsala sa pancreas, tiyan, malaki at maliit na bituka, at atay.
Mga sintomas cystic fibrosis
Ang cystic fibrosis ay nagpapakita ng sarili sa iba't ibang mga klinikal na sintomas. Sa mga bagong silang, ang sakit ay maaaring magpakita mismo sa meconium ileus. Dahil sa kakulangan o kahit na kumpletong kawalan ng trypsin, ang meconium ay nagiging napakasiksik, malapot, at naipon sa ileocecal region. Dagdag pa, ang bituka na sagabal ay bubuo, na kung saan ay ipinahayag sa pamamagitan ng matinding pagsusuka na may isang admixture ng apdo, distension ng tiyan, kakulangan ng meconium excretion, pag-unlad ng mga sintomas ng peritonitis, at mabilis na pagtaas sa mga klinikal na pagpapakita ng malubhang pagkalasing sindrom. Ang bata ay maaaring mamatay sa mga unang araw ng buhay kung ang kagyat na interbensyon sa kirurhiko ay hindi ginanap.
Sa hindi gaanong malubhang mga kaso, ang isang katangian ng pag-sign ng cystic fibrosis ay sagana, madalas na dumi, hindi mabagal, na may malaking halaga ng taba, na may isang napaka hindi kanais-nais na amoy. Sa 1/3 ng mga pasyente, ang prolaps ng tumbong ay sinusunod.
Kasunod nito, ang mga pasyente ay patuloy na nakakaranas ng intestinal dysfunction, malabsorption syndrome, malubhang physical development disorder, at matinding hypovitaminosis.
Sa una o ikalawang taon ng buhay, ang mga sintomas ng pinsala sa bronchopulmonary system (banayad na anyo ng sakit) ay lilitaw, na kung saan ay ipinahayag sa pamamagitan ng isang ubo na maaaring labis na binibigkas at kahawig ng isang ubo na may whooping cough. Ang ubo ay sinamahan ng sianosis, igsi ng paghinga, at ang paghihiwalay ng makapal na plema, sa una ay mauhog, at pagkatapos ay purulent. Unti-unti, nabuo ang isang klinikal na larawan ng talamak na obstructive bronchitis at bronchiectasis, pulmonary emphysema at respiratory failure. Ang mga bata ay lubhang madaling kapitan sa acute respiratory viral at bacterial infections, na nag-aambag sa mga exacerbations at pag-unlad ng bronchopulmonary pathology. Posible ang pag-unlad ng bronchial hika na umaasa sa impeksyon.
Sa mga batang nasa paaralan, ang cystic fibrosis ay maaaring magpakita mismo bilang "intestinal colic". Ang mga pasyente ay nagrereklamo ng matinding paroxysmal na pananakit ng tiyan, bloating, at paulit-ulit na pagsusuka. Kapag palpating ang tiyan, ang mga siksik na pormasyon ay tinutukoy, na matatagpuan sa projection ng malaking bituka - fecal masa na may halong makapal, siksik na uhog. Ang mga bata ay napakahilig sa pagbuo ng hypochloremic alkalosis dahil sa labis na paglabas ng asin na may pawis sa mainit na panahon, habang ang "salt frost" ay lumilitaw sa balat ng bata.
Mga karamdaman sa bronchopulmonary system sa mga matatanda
Ang pinsala sa bronchopulmonary system sa mga pasyente na may cystic fibrosis (pulmonary form ng sakit) ay nailalarawan sa pamamagitan ng pag-unlad ng talamak purulent obstructive bronchitis, bronchiectasis, talamak na pneumonia, pulmonary emphysema, respiratory failure, at pulmonary heart disease. Ang ilang mga pasyente ay nagkakaroon ng pneumothorax at iba pang komplikasyon ng cystic fibrosis: atelectasis, lung abscesses, hemoptysis, pulmonary hemorrhage, at infection-dependent bronchial asthma.
Ang mga pasyente ay nagreklamo ng isang masakit na paroxysmal na ubo na may napakalapot, mahirap paghiwalayin ang mucopurulent na plema, kung minsan ay may isang admixture ng dugo. Bilang karagdagan, ang igsi ng paghinga ay lubhang katangian, una sa panahon ng pisikal na pagsusumikap, at pagkatapos ay sa pahinga. Ang igsi ng paghinga ay sanhi ng bronchial obstruction. Maraming mga pasyente ang nagrereklamo ng talamak na rhinitis na sanhi ng polyposis at sinusitis. Ang makabuluhang kahinaan, progresibong pagbaba sa pagganap, madalas na talamak na respiratory viral disease ay katangian din. Sa pagsusuri, ang pansin ay iginuhit sa maputlang balat, puffiness ng mukha, cyanosis ng mga nakikitang mucous membrane, at matinding igsi ng paghinga. Sa pag-unlad ng decompensated pulmonary heart disease, lumilitaw ang edema sa mga binti. Ang pampalapot ng mga terminal phalanges ng mga daliri sa anyo ng mga drumstick, at mga kuko sa anyo ng mga baso ng relo ay maaaring obserbahan. Ang dibdib ay nagkakaroon ng hugis-barrel na anyo (dahil sa pag-unlad ng pulmonary emphysema).
Ang pagtambulin ng mga baga ay nagpapakita ng mga palatandaan ng emphysema - isang tunog ng kahon, isang matalim na limitasyon ng kadaliang mapakilos ng gilid ng baga, at isang pagbaba ng ibabang hangganan ng mga baga. Ang auscultation ng mga baga ay nagpapakita ng malupit na paghinga na may pinahabang pagbuga, kalat-kalat na tuyo na wheezing, at basa-basa na daluyan at pinong bumubulusok na wheezing. Sa matinding emphysema ng baga, ang paghinga ay humihina nang husto.
Extrapulmonary manifestations ng cystic fibrosis
Ang extrapulmonary manifestations ng cystic fibrosis ay maaaring maging malinaw at madalas mangyari.
Pancreatic pinsala
Ang kakulangan ng exocrine function ng pancreas ng iba't ibang kalubhaan ay sinusunod sa 85% ng mga pasyente na may cystic fibrosis. Sa maliit na pinsala sa pancreas, ang maldigestion at malabsorption syndrome ay wala, mayroon lamang mga pagpapakita ng laboratoryo ng kakulangan sa exocrine (mababang antas ng trypsin at lipase sa dugo at mga nilalaman ng duodenal; madalas na malubhang steatorrhea). Ito ay kilala na upang maiwasan ang maldigestion syndrome, ang pagtatago lamang ng 1 hanggang 2% ng kabuuang lipase ay sapat. Tanging ang mga makabuluhang kaguluhan ng exocrine function ay clinically manifested.
Sa ilalim ng normal na mga kondisyon, ang acini ng pancreas ay gumagawa ng isang likidong pagtatago na mayaman sa mga enzyme. Habang ang pagtatago ay gumagalaw sa kahabaan ng excretory duct, ito ay pinayaman ng tubig at mga anion, at ito ay nagiging mas likido. Sa cystic fibrosis, dahil sa isang disorder sa istraktura at pag-andar ng transmembrane regulator (chloride channel), ang pancreatic secretion ay hindi tumatanggap ng sapat na dami ng likido, ito ay nagiging malapot, at ang bilis ng paggalaw nito kasama ang excretory duct ay bumagal nang husto. Ang mga protina ng pagtatago ay idineposito sa mga dingding ng maliliit na excretory ducts, na nagreresulta sa kanilang sagabal. Sa pag-unlad ng sakit, ang pagkasira at pagkasayang ng acini sa huli ay bubuo - ang talamak na pancreatitis na may exocrine pancreatic insufficiency ay nabuo. Ito ay clinically reflected sa pagbuo ng maldigestion at malabsorption syndromes. Ang pancreatic insufficiency ay ang pangunahing sanhi ng fat malabsorption sa cystic fibrosis, ngunit ito ay karaniwang sinusunod na may makabuluhang kakulangan sa lipase. Ipinapahiwatig ng Forsher at Durie (1991) na sa kumpletong kawalan ng pancreatic lipase, ang taba ay nasira at nasisipsip ng 50-60%, na dahil sa pagkakaroon ng gastric at salivary (sublingual) lipase, ang aktibidad na kung saan ay malapit sa mas mababang limitasyon ng pamantayan. Kasabay ng pagkagambala ng pagkasira at pagsipsip ng taba, mayroong pagkagambala sa pagkasira ng protina at muling pagsipsip. Humigit-kumulang 50% ng protina na natanggap kasama ng pagkain ay nawawala kasama ng mga dumi. Ang pagsipsip ng carbohydrate ay apektado sa isang mas mababang lawak sa kabila ng kakulangan ng α-amylase, ngunit ang metabolismo ng carbohydrate ay maaaring makabuluhang magambala.
Ang pinsala sa pancreas ay ipinahayag sa pagbuo ng maldigestion at malabsorption syndrome na may makabuluhang pagbaba ng timbang at masaganang mataba na dumi.
Ang pag-unlad ng maldigestion at malabsorption syndromes ay pinadali din ng matinding dysfunction ng bituka glandula, may kapansanan na pagtatago ng bituka juice at pagbawas sa nilalaman ng bituka enzymes sa loob nito.
Ang maldigestion at malabsorption syndromes ay tinatawag ding intestinal form ng cystic fibrosis.
Ang kapansanan sa endocrine function ng pancreas (diabetes mellitus) ay sinusunod sa mga pasyente na may cystic fibrosis sa mga huling yugto ng sakit (sa 2% ng mga bata at 15% ng mga matatanda)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Pinsala sa atay at biliary tract
Sa 13% ng mga pasyente na may halo-halong at bituka na anyo ng cystic fibrosis, bubuo ang cirrhosis ng atay. Ito ay pinakakaraniwan para sa mga mutasyon na W128X, delta-P508 at X1303K. Ang biliary cirrhosis ng atay na may portal hypertension ay napansin sa 5-10% ng mga pasyente. Ayon kay Welch, Smith (1995), ang mga klinikal, morphological, laboratoryo, instrumental na mga palatandaan ng pinsala sa atay ay napansin sa 86% ng mga pasyente na may cystic fibrosis.
Maraming mga pasyente na may cystic fibrosis ay nagkakaroon din ng talamak na cholecystitis, kadalasang calculous.
Dysfunction ng mga glandula ng kasarian
Ang mga pasyente na may cystic fibrosis ay maaaring makaranas ng azoospermia, na siyang sanhi ng pagkabaog. Ang pagbabawas ng pagkamayabong ay karaniwan din para sa mga kababaihan.
Mga yugto
Mayroong tatlong antas ng kalubhaan ng pulmonary cystic fibrosis.
Ang banayad na anyo ng cystic fibrosis ay nailalarawan sa pamamagitan ng mga bihirang exacerbations (hindi hihigit sa isang beses sa isang taon); sa mga panahon ng pagpapatawad, ang mga klinikal na pagpapakita ay halos wala, at ang mga pasyente ay maaaring gumana.
Katamtamang kalubhaan - ang mga exacerbations ay sinusunod 2-3 beses sa isang taon at tumatagal ng mga 2 buwan o higit pa. Sa yugto ng exacerbation, mayroong isang matinding ubo na may mahirap na paghiwalayin ang plema, igsi ng paghinga kahit na may menor de edad na pisikal na pagsusumikap, subfebrile na temperatura ng katawan, pangkalahatang kahinaan, pagpapawis. Kasabay nito, mayroong isang paglabag sa exocrine function ng pancreas. Sa yugto ng pagpapatawad, ang kapasidad ng pagtatrabaho ay hindi ganap na naibalik, ang igsi ng paghinga sa panahon ng pisikal na pagsusumikap ay nananatili.
Ang matinding kurso ay nailalarawan sa pamamagitan ng napakadalas na mga exacerbations ng sakit. Ang mga pagpapatawad ay halos wala. Sa klinikal na larawan, malubhang kabiguan sa paghinga, mga sintomas ng talamak na sakit sa baga sa puso, madalas na decompensated, hemoptysis ay tipikal, dumating sa unahan. Ang makabuluhang pagbaba ng timbang ay sinusunod, ang mga pasyente ay ganap na hindi pinagana. Bilang isang patakaran, ang malubhang bronchopulmonary pathology ay sinamahan ng isang matinding ipinahayag na dysfunction ng pancreas.
Mga Form
- Mga sugat sa bronchopulmonary
- Paulit-ulit at paulit-ulit na pulmonya na may matagal na kurso.
- Abscessing pneumonia, lalo na sa mga sanggol.
- Talamak na pulmonya, lalo na bilateral.
- Ang bronchial asthma na matigas ang ulo sa tradisyunal na therapy.
- Paulit-ulit na bronchitis, bronchiolitis, lalo na sa kultura ng Pseudomonas aeruginosa.
- Mga pagbabago sa gastrointestinal tract
- Meconium ileus at mga katumbas nito.
- Syndrome ng may kapansanan na pagsipsip ng bituka ng hindi kilalang genesis.
- Obstructive jaundice sa mga bagong silang na may matagal na kurso.
- Cirrhosis ng atay.
- Diabetes mellitus.
- Gastroesophageal reflux.
- Cholelithiasis.
- Rectal prolapse.
- Mga pagbabago sa ibang mga organo at sistema
- Mga karamdaman sa paglaki at pag-unlad.
- Naantala ang sekswal na pag-unlad.
- Infertility ng lalaki.
- Mga polyp sa ilong.
- Mga kapatid mula sa mga pamilyang may cystic fibrosis.
[ 24 ]
Mga komplikasyon at mga kahihinatnan
Ang mga komplikasyon mula sa gastrointestinal tract ay kinabibilangan ng:
- Ang diabetes mellitus ay bubuo sa 8-12% ng mga pasyente na higit sa 25 taong gulang.
- Fibrosing colonopathy.
- Meconium ileus sa neonatal period (sa 12% ng mga bagong silang na may cystic fibrosis), distal intestinal obstruction syndrome, rectal prolapse, peptic ulcer disease at gastroesophageal reflux disease.
Mga komplikasyon sa atay:
- Sakit sa mataba sa atay (sa 30-60% ng mga pasyente),
- Focal biliary cirrhosis, multinodular biliary cirrhosis, at nauugnay na portal hypertension.
Ang portal hypertension kung minsan ay humahantong sa kamatayan dahil sa esophageal varices.
Ang pagkalat ng cholecystitis at gallstones ay mas mataas sa mga pasyente na may cystic fibrosis kaysa sa ibang mga indibidwal.
Naantala ang pagdadalaga at pagbaba ng fertility at iba pang komplikasyon. Karamihan sa mga lalaki ay may azoospermia at underdevelopment ng vas deferens.
Diagnostics cystic fibrosis
Pangkalahatang pagsusuri ng dugo - tipikal ang anemia na may iba't ibang kalubhaan, karaniwan ay normo- o hypochromic. Ang anemia ay may polyfactorial genesis (nabawasan ang pagsipsip ng iron at bitamina B12 sa bituka dahil sa pagbuo ng malabsorption syndrome). Posible ang leukopenia, na may pag-unlad ng purulent bronchitis at pneumonia - leukocytosis, nadagdagan ang ESR.
Pangkalahatang pagsusuri ng ihi - walang makabuluhang pagbabago, sa mga bihirang kaso ay sinusunod ang bahagyang proteinuria.
Coprological na pagsusuri - steatorrhea, creatorrhea ay sinusunod. Inirerekomenda ni Becker (1987) ang pagsukat ng chymotrypsin at fatty acid sa mga dumi. Bago matukoy ang chymotrypsin sa mga feces, kinakailangan na ihinto ang pagkuha ng digestive enzymes ng hindi bababa sa 3 araw bago ang pagsusuri. Sa cystic fibrosis, ang halaga ng chymotrypsin sa feces ay nabawasan, at ang halaga ng mga fatty acid ay nadagdagan (normal na excretion ng fatty acids ay mas mababa sa 20 mmol / araw). Kinakailangang isaalang-alang na ang pagtaas ng paglabas ng mga fatty acid na may feces ay sinusunod din sa:
- kakulangan ng conjugated fatty acid sa maliit na bituka dahil sa pagkabigo sa atay, sagabal sa mga duct ng apdo, makabuluhang kolonisasyon ng bacterial ng maliit na bituka (sa kasong ito, nangyayari ang masinsinang hydrolysis ng mga acid ng apdo);
- ileitis;
- celiac disease (na may pag-unlad ng malabsorption syndrome);
- enteritis;
- mga lymphoma sa bituka;
- sakit ng whipple;
- allergy sa pagkain;
- pinabilis na transit ng mga masa ng pagkain sa pagtatae ng iba't ibang pinagmulan, carcinoid syndrome, thyrotoxicosis.
Biochemical blood test - nabawasan ang kabuuang antas ng protina at albumin, nadagdagan ang mga antas ng alpha2 at gamma globulins, bilirubin at aminotransferases (sa kaso ng pinsala sa atay), nabawasan ang aktibidad ng amylase, lipase, trypsin at iron at calcium na antas (sa kaso ng pagbuo ng maldigestion syndrome, malabsorption).
Pagsusuri ng plema - pagkakaroon ng isang malaking bilang ng mga neutrophilic leukocytes at microorganisms (sa panahon ng sputum bacterioscopy).
Ang isang pag-aaral ng absorption function ng maliit na bituka at ang exocrine function ng pancreas ay nagpapakita ng mga makabuluhang kaguluhan.
X-ray na pagsusuri sa mga baga - nagpapakita ng mga pagbabago, ang kalubhaan nito ay depende sa kalubhaan at yugto ng sakit. Ang pinaka-katangiang pagbabago ay:
- nadagdagan ang pulmonary patterning dahil sa peribronchial interstitial na pagbabago;
- pagpapalawak ng mga ugat ng baga;
- larawan ng lobular, subsegmental o kahit segmental atelectasis ng baga;
- nadagdagan ang transparency ng mga patlang ng baga, pangunahin sa itaas na mga seksyon, mababang posisyon at hindi sapat na kadaliang mapakilos ng diaphragm, pagpapalawak ng retrosternal space (manifestation ng pulmonary emphysema);
- segmental o polysegmental infiltration ng tissue ng baga (sa pagbuo ng pneumonia).
Ang bronchography ay nagpapakita ng mga pagbabago na dulot ng sagabal ng bronchi sa pamamagitan ng malapot na plema (fragmentation ng pagpuno ng bronchi na may kaibahan, hindi pantay na mga contour, ang kababalaghan ng bronchial rupture, isang makabuluhang pagbaba sa bilang ng mga lateral branch), pati na rin ang mga bronchoecgases (cylindrical, mixed), na naisalokal pangunahin sa mas mababang mga bahagi ng baga.
Ang bronchoscopy ay nagpapakita ng diffuse purulent bronchitis na may masaganang makapal, malapot na plema at fibrinous na mga pelikula.
Spirometry - na sa maagang yugto ng sakit ay nagpapakita ng respiratory failure ng obstructive type (nabawasan ang FVC, FEV1, Tiffno index), mahigpit (nabawasan ang FVC) o, kadalasan, obstructive-restrictive (nabawasan ang FVC, FVC, FEV1, Tiffno index).
Ang Gibson and Cook sweat test (sweat electrolyte test) ay nagsasangkot ng pagpapasigla ng pagpapawis gamit ang pilocarpine electrophoresis na may kasunod na pagtukoy ng mga chlorides sa pawis. Inilalarawan ni Doerehuk (1987) ang pagsusulit bilang mga sumusunod. Ang Pilocarpine electrophoresis ay ginaganap sa bisig, ang electric current ay 3 mA. Matapos linisin ang balat na may distilled water, ang pawis ay kinokolekta gamit ang filter na papel na inilagay sa stimulated na lugar, na natatakpan ng gasa upang maiwasan ang pagsingaw mula dito. Pagkatapos ng 30-60 minuto, ang filter na papel ay aalisin at i-eluted sa distilled water. Ang dami ng pawis na nakolekta ay sinusukat. Upang makakuha ng maaasahang mga resulta, kinakailangan upang mangolekta ng hindi bababa sa 50 mg (mas mabuti 100 mg) ng pawis.
Kung ang konsentrasyon ng chloride ay higit sa 60 mmol/l, ang diagnosis ng cystic fibrosis ay itinuturing na maaaring mangyari; kung ang konsentrasyon ng klorido ay higit sa 100 mmol/l, ito ay maaasahan; sa kasong ito, ang pagkakaiba sa konsentrasyon ng chlorine at sodium ay hindi dapat lumampas sa 8-10 mmol/l. Inirerekomenda ni Hadson (1983) na kung ang nilalaman ng sodium at chloride sa pawis ay borderline, isang prednisolone test ang dapat gawin (5 mg pasalita sa loob ng 2 araw, na sinusundan ng pagpapasiya ng electrolytes sa pawis). Sa mga indibidwal na hindi dumaranas ng cystic fibrosis, ang antas ng sodium sa pawis ay bumababa sa mas mababang limitasyon ng normal; sa cystic fibrosis, hindi ito nagbabago. Inirerekomenda ang pagsusuri sa pawis para sa bawat bata na may talamak na ubo.
Ang pagsusuri ng mga spot ng dugo o mga sample ng DNA para sa mga pangunahing mutasyon ng gene ng cystic fibrosis ay ang pinakasensitibo at partikular na pagsusuri sa diagnostic. Gayunpaman, ang pamamaraang ito ay angkop lamang para sa mga bansa kung saan ang delta-P508 mutation rate ay mas mataas sa 80%. Bilang karagdagan, ang pamamaraan ay napakamahal at kumplikado sa teknikal.
Ang prenatal diagnosis ng cystic fibrosis ay isinasagawa sa pamamagitan ng pagtukoy sa mga isoenzymes ng alkaline phosphatase sa amniotic fluid. Ang pamamaraang ito ay nagiging posible mula sa 18-20 na linggo ng pagbubuntis.
Ang pangunahing pamantayan para sa pag-diagnose ng cystic fibrosis ay ang mga sumusunod:
- mga indikasyon sa anamnesis ng naantala na pisikal na pag-unlad sa pagkabata, paulit-ulit na malalang sakit sa paghinga, dyspeptic disorder at pagtatae, ang pagkakaroon ng cystic fibrosis sa malapit na kamag-anak;
- talamak na obstructive bronchitis, madalas na paulit-ulit, na may pag-unlad ng bronchiectasis at pulmonary emphysema, madalas na paulit-ulit na pneumonia;
- talamak na paulit-ulit na pancreatitis na may markang pagbaba sa exocrine function, malabsorption syndrome;
- nadagdagan ang chlorine content sa pawis ng pasyente;
- kawalan ng katabaan na may napanatili na sekswal na function.
Ang matagumpay na diagnosis at differential diagnosis ng cystic fibrosis ay pinadali sa pamamagitan ng pagtukoy sa mga grupo ng panganib.
Cystic Fibrosis Screening Program
- Pangkalahatang pagsusuri ng dugo, ihi, plema.
- Bacteriological analysis ng plema.
- Pagsusuri ng coprological.
- Pagsusuri sa dugo ng biochemical: pagpapasiya ng kabuuang mga fraction ng protina at protina, glucose, bilirubin, aminotransferases, alkaline phosphatase, gamma-glutamyl transpeptidases, potassium, calcium, iron, lipase, amylase, trypsin.
- Pag-aaral ng exocrine function ng pancreas at ang absorptive function ng bituka.
- Fluoroscopy at radiography ng baga, CT scan ng baga.
- ECG.
- Echocardiography.
- Bronchoscopy at bronchography.
- Spirometry.
- Pagsubok sa pawis.
- Konsultasyon sa isang geneticist.
- Pagsusuri ng mga spot ng dugo o mga sample ng DNA para sa mga pangunahing mutasyon ng cystic fibrosis gene.
Anong mga pagsubok ang kailangan?
Sino ang dapat makipag-ugnay?
Paggamot cystic fibrosis
Ang uri at kalubhaan ng mga sintomas ng cystic fibrosis ay maaaring mag-iba nang malaki, kaya walang tipikal na plano ng paggamot; ito ay indibidwal sa bawat indibidwal.
Ang therapy ay binubuo ng mga sumusunod na therapeutic measure:
- Ang mga ehersisyo sa paghinga at postural drainage ay nakakatulong upang maalis ang makapal na uhog na naipon sa mga baga. Ang ilang mga pamamaraan sa paglilinis ng daanan ng hangin ay nangangailangan ng tulong mula sa mga miyembro ng pamilya, kaibigan, o isang pulmonologist. Maraming tao ang gumagamit ng inflatable chest vest na nagvibrate sa mataas na frequency.

- Mga gamot sa paglanghap na mayroong bronchodilator, draining (mucolytics) at antibacterial effect (halimbawa, fluoroquinolones).
- Mga paghahanda na naglalaman ng pancreatic enzymes upang mapabuti ang panunaw. Ang mga paghahandang ito ay kinukuha sa panahon ng pagkain.
- Mga multivitamin (kabilang ang mga bitamina na natutunaw sa taba).
Noong 2015, inaprubahan ng FDA ang pangalawang gamot para gamutin ang cystic fibrosis na nagta-target ng may sira na protina na kilala bilang CFTR. Ang unang gamot, isang tinatawag na CFTR modulator, ay naaprubahan noong 2012. Ang mga modulator ng CFTR ay inaasahang magpapahaba ng buhay ng ilang taong may cystic fibrosis nang mga dekada.
Maaaring kailanganin ang operasyon upang gamutin ang mga sumusunod na komplikasyon sa paghinga:
- Pneumothorax, napakalaking paulit-ulit o paulit-ulit na hemoptysis, nasal polyp, paulit-ulit at talamak na sinusitis.
- Meconium ileus, intussusception, rectal prolaps.
Ang paglipat ng baga ay isinasagawa sa terminal stage ng sakit.
Pagtataya
Ang average na edad ng kaligtasan ng buhay ng mga pasyente na may cystic fibrosis ay mula 35 hanggang 40 taon. Ang average na edad ng kaligtasan ng buhay ay mas mataas sa mga lalaki kaysa sa mga babae.
Gamit ang mga modernong diskarte sa paggamot, 80% ng mga pasyente ay umabot sa pagtanda. Gayunpaman, ang cystic fibrosis ay makabuluhang nililimitahan ang mga kakayahan ng pasyente. Wala pa ring lunas sa sakit na ito.