Medikal na dalubhasa ng artikulo

Mga bagong publikasyon
Prion - mga ahente ng sanhi ng mga sakit sa prion
Huling nasuri: 06.07.2025

Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Ang mabagal na impeksyon sa viral ay nailalarawan sa pamamagitan ng mga espesyal na pamantayan:
- isang hindi karaniwang mahabang panahon ng pagpapapisa ng itlog (buwan, taon);
- isang tiyak na sugat ng mga organo at tisyu, lalo na ang central nervous system;
- mabagal, matatag na pag-unlad ng sakit;
- hindi maiiwasang nakamamatay na kinalabasan.

Ang ilang mga pathogen na nagdudulot ng talamak na impeksyon sa viral ay maaari ding maging sanhi ng mabagal na impeksyon sa viral. Halimbawa, ang virus ng tigdas kung minsan ay nagdudulot ng SSPE, at ang rubella virus ay nagdudulot ng progresibong congenital rubella at rubella panencephalitis.
Ang isang tipikal na mabagal na impeksyon sa viral ng mga hayop ay sanhi ng visna/madi virus, na isang retrovirus. Ito ang sanhi ng ahente ng mabagal na impeksyon sa viral at progresibong pneumonia sa mga tupa. Ang puting bagay ng utak ay nawasak, ang paralisis ay bubuo (visna - pag-aaksaya); ang talamak na pamamaga ng mga baga at pali ay nangyayari.
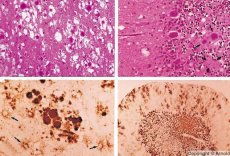
Ang mga sakit na katulad ng kanilang mga tampok sa pagbagal ng mga impeksyon sa virus ay sanhi ng mga prion - ang mga sanhi ng mga impeksyon sa prion. Ang mga sakit sa prion ay isang pangkat ng mga progresibong karamdaman ng central nervous system ng mga tao at hayop. Sa mga tao, ang pag-andar ng central nervous system ay may kapansanan, ang mga pagbabago sa personalidad ay nangyayari, at ang mga karamdaman sa paggalaw ay nangyayari. Ang mga sintomas ng sakit ay karaniwang tumatagal mula sa ilang buwan hanggang ilang taon, na nagtatapos sa kamatayan. Noong nakaraan, ang mga impeksyon sa prion ay isinasaalang-alang kasama ang tinatawag na mga ahente ng sanhi ng mabagal na impeksyon sa viral.
Ang ilang mga ahente na nagdudulot ng mga sakit sa prion ay unang naiipon sa mga lymphoid tissue. Ang mga prion, na pumapasok sa utak, ay naipon sa malalaking dami, na nagiging sanhi ng amyloidosis (extracellular dysproteinosis, na nailalarawan sa pamamagitan ng pagtitiwalag ng amyloid na may pag-unlad ng pagkasayang at sclerosis ng tissue) at astrocytosis (paglaganap ng astrocytic neuroglia, hyperproduction ng glial fibers). Nabubuo ang mga fibril, pinagsama-samang protina o amyloid at spongiform sa utak (transmissible spongiform encephalopathies). Bilang isang resulta, ang mga pagbabago sa pag-uugali, ang koordinasyon ng mga paggalaw ay may kapansanan, ang pagkapagod na may nakamamatay na kinalabasan ay bubuo. Ang kaligtasan sa sakit ay hindi nabuo. Ang mga sakit sa prion ay mga conformational na sakit na nabubuo bilang resulta ng hindi tamang pagtiklop (paglabag sa tamang conform) ng cellular protein na kinakailangan para sa normal na paggana ng katawan. Ang mga ruta ng paghahatid ng prion ay iba-iba:
- ruta ng pagkain - mga nahawaang produkto ng pinagmulan ng hayop, mga additives ng pagkain mula sa mga hilaw na organo ng baka, atbp.:
- paghahatid sa pamamagitan ng pagsasalin ng dugo, pangangasiwa ng mga gamot na pinagmulan ng hayop, paglipat ng organ at tissue, paggamit ng mga nahawaang instrumento sa operasyon at ngipin;
- paghahatid sa pamamagitan ng immunobiological paghahanda (impeksyon ng 1500 tupa na may PrP''' sa pamamagitan ng brain formol vaccine mula sa may sakit na tupa ay kilala).
Ang mga pathological prion, na pumasok sa bituka, ay dinadala sa dugo at lymph. Pagkatapos ng peripheral replication sa spleen, appendix, tonsil at iba pang lymphoid tissues, inililipat sila sa utak sa pamamagitan ng peripheral nerves (neuroinvasion). Ang direktang pagtagos ng mga prion sa utak sa pamamagitan ng blood-brain barrier ay posible. Noong nakaraan, pinaniniwalaan na ang gitnang sistema ng nerbiyos ay ang tanging tisyu kung saan naipon ang mga pathological prion, ngunit lumitaw ang mga pag-aaral na nagbago sa hypothesis na ito. Ito ay lumabas na ang akumulasyon ng mga prion sa pali ay nauugnay sa pagtaas at paggana ng mga follicular dendritic cells.
 [
[ Mga katangian ng prion
Ang normal na cellular isoform ng prion protein na may molekular na timbang na 33-35 kDa ay tinutukoy ng prion protein gene (ang prion gene - PrNP ay matatagpuan sa ika-20 na kromosoma ng tao). Lumilitaw ang normal na gene sa ibabaw ng cell (naka-angkla sa lamad ng glycoprotein ng molekula), sensitibo sa protease. Kinokontrol nito ang paghahatid ng mga nerve impulses, pang-araw-araw na cycle, proseso ng oksihenasyon, nakikilahok sa metabolismo ng tanso sa central nervous system at sa regulasyon ng bone marrow stem cell division. Bilang karagdagan, ang prion gene ay matatagpuan sa spleen, lymph nodes, balat, gastrointestinal tract at follicular dendritic cells.
Paglaganap ng mga pathological prion
Ang pagbabago ng prion sa mga binagong anyo ay nangyayari kapag ang kinetically controlled equilibrium sa pagitan ng mga ito ay nagambala. Ang proseso ay pinahusay ng isang pagtaas sa dami ng pathological (PrP) o exogenous prion. Ang PrP ay isang normal na protina na naka-angkla sa lamad ng cell. Ang PrP' ay isang globular hydrophobic protein na bumubuo ng mga pinagsama-samang kasama nito at PrP'' sa ibabaw ng cell: bilang resulta, ang PrP' ay binago sa PrP'' at pagkatapos ay nagpapatuloy ang cycle. Ang pathological na anyo ng PrP''' ay naipon sa mga neuron, na nagbibigay sa cell ng isang spongy na hitsura.
Kuru
Ang sakit na prion, na dati ay karaniwan sa mga Papuan (nangangahulugang nanginginig o nanginginig) sa silangang bahagi ng isla ng New Guinea. Ang mga nakakahawang katangian ng sakit ay napatunayan ni K. Gajdusek. Ang pathogen ay ipinapadala sa pamamagitan ng pagkain bilang isang resulta ng ritwal na cannibalism - pagkain ng hindi sapat na luto, prion-infected na utak ng mga patay na kamag-anak. Bilang resulta ng pinsala sa gitnang sistema ng nerbiyos, ang paggalaw at lakad ay may kapansanan, ang panginginig at euphoria ("pagtatawanan na kamatayan") ay lumilitaw. Ang panahon ng pagpapapisa ng itlog ay tumatagal ng 5-30 taon. Ang pasyente ay namatay pagkatapos ng isang taon.
Sakit na Creutzfeldt-Jakob
Prion disease, na nagpapakita ng sarili bilang dementia, visual at cerebellar disorder at mga sakit sa paggalaw na may nakamamatay na kinalabasan pagkatapos ng 4-5 na buwan ng pagkakasakit sa klasikong variant ng Creutzfeldt-Jakob disease at pagkatapos ng (3-14 na buwan sa bagong variant ng Creutzfeldt-Jakob disease. Ang incubation period ay maaaring umabot ng 20 taon. Iba't ibang mga ruta ng impeksiyon at: ng mga posibleng sanhi ng sakit.
- kapag kumakain ng hindi sapat na heat-treated na mga produktong hayop, tulad ng karne at utak mula sa mga baka na may bovine spongiform encephalopathy;
- sa panahon ng paglipat ng tisyu, tulad ng paglipat ng kornea, pagsasalin ng dugo, paggamit ng mga hormone at iba pang biologically active substance na pinagmulan ng hayop, paggamit ng catgut, kontaminado o hindi sapat na isterilisadong mga instrumento sa pag-opera, mga manipulasyon ng prosectoral;
- sa kaso ng hyperproduction ng PrR at iba pang mga kondisyon na nagpapasigla sa proseso ng pag-convert ng PrR' sa PrR".
Ang sakit ay maaari ring bumuo bilang isang resulta ng isang mutation o pagpasok sa rehiyon ng prion gene. Ang kalikasan ng pamilya ng sakit ay karaniwan dahil sa genetic predisposition sa Creutzfeldt-Jakob disease. Sa bagong variant ng sakit na Creutzfeldt-Jakob, nagkakaroon ng mga karamdaman sa mas bata na edad (average na edad 28 taon), sa kaibahan sa klasikong variant (average na edad 65 taon). Sa bagong variant ng sakit na Creutzfeldt-Jakob, ang abnormal na protina ng prion ay naipon hindi lamang sa gitnang sistema ng nerbiyos, kundi pati na rin sa mga lymphoreticular na tisyu, kabilang ang mga tonsils.
Gerstmann-Sträussler-Scheinker syndrome
Hereditary prion disease, sinamahan ng demensya, hypotonia, swallowing disorder (dysphagia), dysarthria. Kadalasan ay may likas na pamilya. Ang panahon ng pagpapapisa ng itlog ay mula 5 hanggang 30 taon. Ang sakit ay nangyayari sa 50-60 taon, ang tagal nito ay mula 5 hanggang 13 taon.
Hereditary fatal insomnia
Isang autoimmune disease na may progresibong insomnia, sympathetic hyperreactivity (hypertension, hyperthermia, hyperhidrosis, tachycardia), panginginig, ataxia, multiclone, guni-guni. Ang pagtulog ay malubhang nagambala. Ang kamatayan ay nangyayari sa pag-unlad ng cardiovascular failure.
Siskisan
Ang Scrapie (mula sa English scrape - to scrape) ay isang sakit na prion ng mga tupa at kambing (scabies), na nangyayari na may pinsala sa central nervous system, mga progresibong sakit sa paggalaw, matinding pangangati ng balat (scabies) at nagtatapos sa pagkamatay ng hayop.
Bovine spongiform encephalopathy
Isang sakit ng mga baka na nailalarawan sa pamamagitan ng pinsala sa gitnang sistema ng nerbiyos, may kapansanan sa koordinasyon ng mga paggalaw at hindi maiiwasang pagkamatay ng hayop. Ang epidemya ng sakit ay unang sumiklab sa Great Britain. Ito ay nauugnay sa pagpapakain ng mga hayop na may karne at buto na naglalaman ng mga pathological prion. Ang panahon ng pagpapapisa ng itlog ay mula 1.5 hanggang 15 taon. Ang utak, spinal cord at eyeballs ng mga hayop ay pinaka-impeksyon.
Mga diagnostic sa laboratoryo ng mga sakit sa prion
Sa panahon ng mga diagnostic, ang mga pagbabago sa spongiform sa utak, astrocytosis (gliosis), at ang kawalan ng mga inflammatory infiltrates ay nabanggit. Ang utak ay nabahiran ng amyloid. Ang mga marker ng protina ng mga sakit sa utak ng prion ay nakita sa cerebrospinal fluid (gamit ang ELISA). Ginagawa ang genetic analysis ng prion gene (PCR).
Pag-iwas sa mga sakit sa prion
Ang autoclaving (sa 134°C sa loob ng 18 min; sa 121°C sa loob ng 1 h), pagsusunog, karagdagang paggamot na may bleach at isang normal na NaCl solution sa loob ng 1 h ay inirerekomenda para sa decontamination ng mga instrumento at mga bagay sa kapaligiran. Para sa hindi tiyak na prophylaxis, ang mga paghihigpit ay ipinakilala sa paggamit ng mga produktong panggamot na pinagmulan ng hayop at ang paggawa ng mga pituitary hormone na pinagmulan ng hayop ay ipinagbabawal. Ang paglipat ng dura mater ay pinaghihigpitan. Ang mga guwantes na goma ay ginagamit kapag nagtatrabaho sa mga likidong diyalogo ng mga pasyente.